АНЦА-ассоциированные интерстициальные заболевания легких: актуальные вопросы диагностики и лечения
В течение последних лет появляются данные о связи интерстициальных заболеваний легких (ИЗЛ) и АНЦА-ассоциированных васкулитов (ААВ), особенно у пациентов с микроскопическим полиангиитом с наличием антител к миелопероксидазе (МПО-АНЦА). В части случаев ИЗЛ является единственным или ведущим проявлением заболевания, определяющим объем терапии и прогноз жизни больного с ААВ. В статье обсуждаются современные концепции патогенеза, подходы к диагностике и лечению АНЦА-ассоциированных ИЗЛ, а также дальнейшие направления изучения данной аутоиммунной патологии.
Интерстициальные заболевания легких (ИЗЛ) характеризуются развитием про грессирующего диффузного воспалительного и/или фиброзирующего поражения легких и сходными клиникорентгенологическими проявлениями и гистологической картиной. В соответствии с классификацией, принятой Американским торакальным и Европейским респираторным обществами в 2013 г., к ИЗЛ относят более 200 заболеваний, как с установленной этиологией, так и идиопатических [1].
Антинейтрофильные цитоплазматические антитела (АНЦА) являются серологическим маркером АНЦА-ассоциированных васкулитов (ААВ), в том числе микроскопического полиангиита (МПА), гранулематоза с полиангиитом (ГПА) и эозинофильного гранулематоза с полиангиитом (ЭГПА) [2]. Поражение легких – одно из частых проявлений ААВ [3]: у 85-90% пациентов ГПА 5 и 25-55% с МПА 7 имеются отклонения при компьютерной томографии органов грудной клетки (КТ). При ЭГПА клинически значимые изменения в легких (за исключением бронхиальной астмы) встречаются реже [10]. Наиболее клинически значимыми вариантами поражения легких при ААВ, ассоциированными с неблагоприятным исходом, можно считать диффузное альвеолярное кровотечение (ДАК) и узловые образования различных размеров, в том числе с зонами распада [11].
В течение нескольких лет появляются данные о связи ИЗЛ и ААВ, особенно ассоциированным с антителами к миелопероксидазе (МПО-АНЦА) [11]. В части случаев при ААВ наблюдается изолированное поражение легких; кроме того, у небольшой части пациентов с идиопатическими интерстициальными пневмониями наличие АНЦА (чаще МПО-АНЦА, чем антитела к протеиназе-3 (ПР3-АНЦА)) не сопровождается симптомами системного васкулита или предшествует его развитию [12].
В настоящее время имеется ограниченное количество информации относительно подходов к диагностике и лечению АНЦА-ассоциированных интерстициальных заболеваний легких (АНЦА-ИЗЛ), что объясняется их относительной редкостью [12]. В то же время, наличие интерстициального поражения легких при ААВ может негативно влиять на прогноз заболевания и повышать риск летального исхода, что подчеркивает необходимость поиска его оптимального лечения. В статье представлен современный взгляд на классификацию АНЦА-ИЗЛ, а также обсуждаются вопросы патогенеза, диагностики, лечения и направления дальнейшего изучения данной патологии.
Эпидемиология и классификация
ИЗЛ встречаются чаще у пациентов с МПА (до 45%) и реже у пациентов с ГПА (менее 5%); при ЭГПА описаны лишь единичные случаи развития интерстициального поражения легких [13,14]. При АНЦА-ИЗЛ отмечается значительное преобладание МПО-АНЦА (46-71%) по сравнению с ПР3АНЦА (0-29%) [13,15-20].
Распространенность ИЗЛ при ААВ зависит от географического региона: в Европе она составляет 2-3%, что существенно ниже по сравнению со странами Азии (2839%) [21,22]. Это может быть отчасти связано с большей частотой выявления МПО-АНЦА в азиатской популяции [11].
Возрастная структура АНЦА-ИЗЛ несколько отличается от таковой в общей группе ААВ. АНЦА-ИЗЛ, как правило, развиваются у пациентов старше 65 лет, хотя единичные случаи описаны даже у детей [11]. В ряде исследований средний возраст на момент диагностики МПА-ассоциированного ИЗЛ (МПА-ИЗЛ) превышал таковой для общей когорты пациентов с МПА (66 и 55 лет, соответственно) и был сопоставим с возрастом пациентов в дебюте идиопатического легочного фиброза (ИЛФ) [7,8]. В нескольких сериях клинических случаев МПА-ИЗЛ наблюдалось незначительное преобладание мужчин (60-65%), однако это отмечалось не во всех наблюдениях [15,23]. Многофакторный анализ в одном из исследований, включавших 62 пациента с ААВ-ИЗЛ, показал, что мужской пол и возраст старше 65 лет были независимыми предикторами развития легочного поражения [24].
Среди АНЦА-ИЗЛ можно выделить три основные группы. Первая включает пациентов с развернутой клинической картиной МПА, развившейся до поражения легких (8-21%); во вторую входят пациенты с ИЗЛ, дебютировавшим одновременно с системным васкулитом (36-67%); третья объединяет АНЦА-позитивных пациентов с ИЗЛ без признаков поражения других органов (14-85%) [11,17,25]. Последний клинический сценарий представляет особый диагностический интерес в связи с возможностью развития в последующем ААВ в срок от нескольких месяцев до 12 лет [11]. В одном исследовании на момент установления диагноза ИЛФ у 4,0% из 504 пациентов определялись МПОАНЦА, у 3,2% – ПР3-АНЦА. В течение последующих 5 лет сероконверсия произошла еще у 11% исходно АНЦА-негативных пациентов, а у одной четверти больных с МПО-АНЦА развился МПА [25]. Наличие других типов аутоантител, в частности ревматоидного фактора (РФ), повышение СОЭ >40 мм/ч, эозинофилия лаважной жидкости и большой объем поражения легочной ткани по данным КТ служили прогностическими факторами АНЦА-сероконверсии [26].
Патогенез
К настоящему времени предложены несколько потенциальных механизмов развития АНЦА-ИЗЛ. Согласно одной из гипотез, формирование интерстициального легочного фиброза при ААВ может быть следствием рецидивирующих эпизодов ДАК [27]. В пользу данной концепции свидетельствует наличие гистологических признаков острого и/или хронического кровотечения более чем у половины пациентов с АНЦА-ИЗЛ [28]. Важно отметить, что у большинства из них отсутствуют анамнестические данные о ДАК, что указывает на вероятное развитие субклинических эпизодов кровотечений [28,29]. Еще одним подтверждением данной гипотезы являются обнаружение в ткани легких значительного количества сидерофагов – гистологических маркеров хронического альвеолярного кровотечения – у пациентов с АНЦА-ИЗЛ и отсутствие данного признака у пациентов с ИЗЛ в рамках других системных аутоиммунных заболеваний, таких как воспалительные миопатии и системная склеродермия [30].
Согласно второй гипотезе, МПО-АНЦА (но не ПР3АНЦА) могут сами по себе играть роль в патогенезе ИЗЛ [31]. В одном исследовании было показано, что активация нейтрофилов антителами к МПО приводила к выработке большого количества окислительных соединений. К ним, в частности, относится гипохлорит-анион, способный стимулировать пролиферацию фибробластов in vitro. Кроме того, МПО-АНЦА могут способствовать повреждению легочной ткани путем локального высвобождения протеолитических ферментов активированными нейтрофилами. Некоторые из этих ферментов, в частности эластаза, могут вызывать развитие легочного фиброза в опытах на животных [32].
Повреждение, вызванное внеклеточными нейтрофильными ловушками, выделяющимися АНЦА-активированными нейтрофилами при их гибели путем нетоза, также может также вносить вклад в легочный патологический процесс. Внеклеточные нейтрофильные ловушки обладают способностью активировать легочные фибробласты и стимулировать их дифференцировку в миофибробласты – один из основных типов клеток, активно вовлеченных в развитие интерстициального легочного фиброза [33].
Еще одна концепция патогенеза АНЦА-ИЗЛ согласуется с фактом развития ИЗЛ до дебюта ААВ и основывается на том, что наличие ИЗЛ само по себе предрасполагает к появления МПО-АНЦА [24]. У пациентов с ИЛФ в лаважной жидкости в большинстве случаев увеличено количество нейтрофилов [34]. При активации нейтрофилы начинают экспрессировать МПО на своей плазматической мембране, что при наличии тканевого воспаления может запускать развитие аутоиммунной реакции и приводить к секреции МПО-АНЦА, а затем и развитию ААВ. В пользу данной гипотезы свидетельствуют появление МПО-АНЦА и развитие МПА у части пациентов с ИЛФ. В этой связи представляет интерес тот факт, что при ААВ, как и при ИЛФ, одним из факторов риска развития ИЗЛ является курение (частота его составила 59% и 23% у пациентов с МПА, у которых отмечалось и отсутствовало ИЗЛ, соответственно) [25].
В то же время, несмотря на ряд сходств, некоторые механизмы развития ИЛФ не играют значимой роли в патогенезе поражения легких при ААВ. В частности, ключевая роль апоптоза альвеолярных эпителиоцитов вследствие длительного воздействия повреждающих факторов, а также эпителиально-мезенхимального перехода не была подтверждена при АНЦА-ассоциированном интерстициальном легочном фиброзе. И наоборот, роль нейтрофилов и системы комплемента в развитии ИЛФ представляется незначительной по сравнению с таковой при АНЦА-ИЗЛ [35].
Клиническаякартина
Основными симптомами у пациентов с АНЦА-ИЗЛ является одышка (50-73%) и малопродуктивный кашель (21-60%) [36,37]. Кровохарканье (5%) и конституциональные проявления, такие как лихорадка (31%) и снижение массы тела (5%), наблюдаются реже [36]. В ранее опубликованных исследованиях не было выявлено достоверной корреляции между титром АНЦА и тяжестью ИЗЛ [36]. В ряде наблюдений клинические проявления у пациентов с фиброзирующими ИЗЛ и наличием АНЦА не отличались от таковых при ИЛФ [36,37].
В то же время, у пациентов с МПА-ИЗЛ в дебюте обычно преобладают конституциональные симптомы (около 80% случаев) – недомогание (31-63%), лихорадка (52-90%) и снижение массы тела (52-58%), а также внелегочные проявления основного заболевания (70100%) [38,39]. Легочные симптомы включают в себя прогрессирующую одышку (30-100%), кровохарканье (21-49%) и кашель (23-84%) [29,38,39]. Следует отметить, что у пациентов с МПА-ИЗЛ реже выявляют признаки системного воспаления, что выражается в более низком уровне СОЭ, более высоком уровне гемоглобина, и, что важно, в меньшей частоте таких проявлений васкулита, как ДАК, поражение периферической нервной системы и почек [29,40].
Диагностика
Повышение уровня СОЭ и содержания С-реактивного белка (СРБ) в дебюте заболевания у пациентов с МПАИЗЛ отмечается в 95% и 73-79% случаев, соответственно [17]. Более чем у 60% из них выявляют также изменения в общем анализе мочи [25]. В то же время, значительное повышение уровня воспалительных маркеров редко встречается при изолированном АНЦАИЗЛ. При сравнении данной группы с ИЛФ в большинстве исследований различий по уровню СРБ не обнаружено [36,37]. При исследовании сывороточных маркеров легочного повреждения у пациентов с ААВ повышение уровня Krebs von der Lungen (KL)-6 ассоциировалось с наличием ИЗЛ [41]. При ААВ-ИЗЛ отмечается четкая корреляция между наличием МПО-АНЦА и ИЗЛ; у пациентов с ПР3-АНЦА развитие ИЗЛ наблюдается значительно реже [24]. Кроме того при наличии ПР3-АНЦА у пациентов с ИЛФ ни в одном случае не было отмечено развития ААВ [25].
При АНЦА-ИЗЛ чаще всего развиваются вентиляционные нарушения рестриктивного типа – снижение форсированной жизненной емкости легких (ФЖЕЛ) и диффузионной способности легких (DLCO), хотя примерно в трети случаев наблюдается умеренная бронхообструкция [15,36,38]. В динамике отмечается тенденция к дальнейшему снижению вентиляционных параметров по мере прогрессирования ИЗЛ. В одном исследовании у больных с ААВ-ИЗЛ в течение 5 лет отмечалось снижение жизненной емкости легких и (ЖЕЛ) и DLCO на 23% и 46%, соответственно, по сравнению с исходными значениями [42].
При исследовании бронхоальвеолярной лаважной жидкости (БАЛЖ) у пациентов с АНЦА-ИЗЛ выявляют нейтрофилию (40-87%), реже – лимфоцитоз и эозинофилию (20% и 26%, соответственно) [15,36]. Признаки острого или хронического альвеолярного кровотечения обнаруживают в половине случаев [15]. По данным ряда авторов, при АНЦА-ИЗЛ количество нейтрофилов и эозинофилов в БАЛЖ было выше, чем при ИЛФ, однако эти результаты подтверждаются не всеми исследованиями [15,37,43].
Основными изменениями на КТ, ассоциированными с ААВ, являются уплотнение по типу «матового стекла» (23-100%), ретикулярные изменения (41-77%), утолщение междолькового интерстиция (41-71%), зоны консолидации (0-78%) и «сотового легкого» (23-100%). Реже выявляют узловые образования в паренхиме (0-45%) и кисты (27%) [22,39,42]. Может наблюдаться поражение дыхательных путей в форме бронхиолита (55%), утолщения стенок бронхов (44%) и бронхоэктазов (32100%). У большинства пациентов (50-100%) изменения в легких носят симметричный характер [38,42,44].
Согласно международным классификационным критериям идиопатических интерстициальных пневмоний, наиболее частым КТ-паттерном ААВ-ИЗЛ, как при наличии МПО-АНЦА, так и ПР3-АНЦА, является обычная интерстициальная пневмония (ОИП; 38-63%), характеризующаяся ретикулярными изменениями, преимущественно в задних и нижних отделах легких, в сочетании с формированием зон «сотового легкого» и уменьшением объема нижних долей. Несколько реже отмечается неспецифическая интерстициальная пневмония (НСИП; 7-31%) и десквамативная интерстициальная пневмония (14%) (рис. 1). У части пациентов (преимущественно курящих мужчин) описано развитие интерстициального фиброза в сочетании с эмфиземой легких (до 21%) [6,38,39].
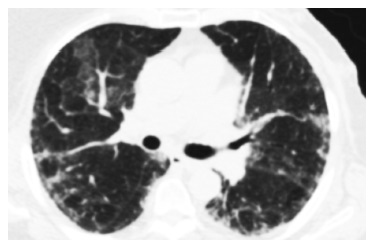 Рис.1. КТ-паттерн НСИП поражения легких у пациентки с МПА. В периферических отделах обоих легких выявляется утолщение междолькового и внутридолькового интерстиция, в сочетании с уплотнение легочной ткани по типу «матового стекла». Обращает на себя внимание относительная сохранность непосредственно субплевральных отделов легких (subpleural sparing).
Рис.1. КТ-паттерн НСИП поражения легких у пациентки с МПА. В периферических отделах обоих легких выявляется утолщение междолькового и внутридолькового интерстиция, в сочетании с уплотнение легочной ткани по типу «матового стекла». Обращает на себя внимание относительная сохранность непосредственно субплевральных отделов легких (subpleural sparing).
Следует отметить, что в 4-40% случаев изменения в легких при АНЦА-ИЗЛ не соответствуют какому-либо конкретному паттерну интерстициальной пневмонии [36]. В спорных случаях для определения показаний к иммуносупрессивной терапии и ее объема требуется гистологическое подтверждения диагноза. В связи с невозможностью проведения видео-ассоциированной торакоскопической биопсии легкого у части пациентов из-за выраженных вентиляционных нарушений и дыхательной недостаточности может быть также рассмотрено выполнение криобиопсии легкого [24].
Как и в случае рентгенологической картины, основным гистологическим паттерном АНЦА-ИЗЛ является ОИП (46-100%), на втором месте по частоте находится НСИП (7-31%) [36,37]. Следует отметить, что несмотря на невысокую распространенность НСИП, в части случаев при основном паттерне ОИП у пациентов также выявляли отдельные зоны с НСИП-подобной гистологической картиной. Кроме того, в отличие от ОИПИЛФ, при АНЦА-ОИП чаще наблюдаются признаки интерстициального воспаления, поражения мелких дыхательных путей, а также лимфоидные фолликулы [36]. Интересно, что признаки активного васкулита редко определяются в биоптатах легкого у пациентов с АНЦА-ИЗЛ [37].
ИЗЛ при ААВ следует дифференцировать с ДАК – одним из наиболее опасных проявлений системного васкулита [11]. Для КТ-картины ДАК в острой стадии характерно наличие диффузно расположенных зон матового стекла, которые в ряде случаев могут занимать большую часть объема легочной паренхимы, с визуализирующимися заполненными сегментарными и субсегментарными бронхами. Скопления крови в полости альвеол могут также формировать различные по размеру зоны консолидации. В подостром периоде (через 2-3 дня после эпизода кровотечения) для ДАК характерно утолщение междолькового и внутридолькового интерстиция, в ряде случаев – на фоне сохраняющегося уплотнения по типу матового стекла (КТ-симптом «булыжной мостовой»). В исходе ДАК в легких могут формироваться множественные нечеткие центрилобулярные очаги, соответствующие интраальвеолярным скоплениям сидерофагов; вследствие обильных рецидивирующих ДАК может развиться грубый интерстициальный легочный фиброз [6,11].
С учетом активной комбинированной иммуносупрессивной терапии, проводимой большинству пациентов с ААВ, в круг дифференциального диагноза следует включать инфекционные осложнения, в первую очередь, обусловленные оппортунистическими микроорганизмами. Так, пневмония, вызванная Pneumocystis jiroveci, характеризуется появлением обширных зон матового стекла в легочной ткани и может имитировать обострение ИЗЛ (рис. 2) [45].
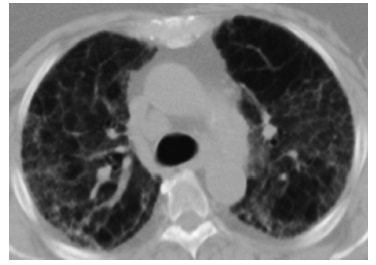 Рис. 2. Прогрессирующие интерстициальные изменения в легких в виде нарастания ретикулярных изменений и увеличения площади «матового стекла» у пациентки с МПА, получающей комбинированную иммуносупрессивную терапию. В периферических отделах обоих легких выявляется утолщение междолькового и внутридолькового интерстиция, в сочетании с уплотнение легочной ткани по типу «матового стекла». Обращает на себя внимание относительная сохранность непосредственно субплевральных отделов легких (subpleural sparing).
Рис. 2. Прогрессирующие интерстициальные изменения в легких в виде нарастания ретикулярных изменений и увеличения площади «матового стекла» у пациентки с МПА, получающей комбинированную иммуносупрессивную терапию. В периферических отделах обоих легких выявляется утолщение междолькового и внутридолькового интерстиция, в сочетании с уплотнение легочной ткани по типу «матового стекла». Обращает на себя внимание относительная сохранность непосредственно субплевральных отделов легких (subpleural sparing).
Лекарственное поражение легких, особенно при длительном приеме и высокой кумулятивной дозе цитостатических препаратов, также должно рассматриваться в качестве причины развития ИЗЛ у пациентов с ААВ [11].
Лечение и прогноз
Согласно современным клиническим рекомендациям, лечение ААВ включает индукцию ремиссии с использованием высокоактивных иммуносупрессивных препаратов (высоких доз ГКС в сочетании с циклофосфамидом или ритуксимабом) с последующим переходом на прием поддерживающей терапии для предотвращения рецидива [46]. В то же время, стандартные протоколы терапии не учитывают особенности клинической картины заболевания в отдельных подгруппах пациентов, в том числе при АНЦА-ИЗЛ. Использование иммуносупрессивной терапии при интерстициальном поражении легких в рамках ААВ отчасти основывается на ее доказанной эффективности при ААВ и ИЗЛ, ассоциированных с рядом системных заболеваний соединительной ткани, в частности системной склеродермией и воспалительными миопатиями [11].
Тем не менее, полученные к настоящему моменту данные об эффективности иммуносупрессивной терапии при лечении ААВ-ИЗЛ носят противоречивый характер. С одной стороны, в ряде работ была выявлена клиническая или рентгенологическая положительная динамика у 80% пациентов с ААВ-ИЗЛ, получавших иммуносупрессивную терапию [36]. В исследовании С. Comarmond и соавт. 3-летняя выживаемость пациентов, получавших комбинированную терапию ГКС в сочетании с циклофосфамидом, составила 94% и превышала таковую у больных, которым проводилась монотерапия глюкокортикостероидами – 65% [17]. Имеются указания на зависимость эффективности иммуносупрессивной терапии от паттерна легочного поражения: у пациентов с КТ-картиной НСИП чаще отмечается положительная динамика легочного поражения в ответ на иммуносупрессивную терапию, тогда как у пациентов с ОИП чаще наблюдается прогрессирование ИЗЛ, несмотря на проводимое лечение, что позволяет провести аналогию с ИЛФ [11].
Нерешенным остается вопрос об эффективности иммуносупрессивной терапии при изолированных АНЦА-ИЗЛ, так как большая часть информации была получена из ретроспективных исследований в неоднородных выборках [12]. В ряде исследований было показано, что прогноз при АНЦА-ИЗЛ достоверно не отличается от такового при ИЛФ (средняя выживаемость – 2,5 и 3,5 года с момента установления диагноза, соответственно), вне зависимости от объема проводимой иммуносупрессивной терапии [47]. И наоборот, другие работы продемонстрировали, что выживаемость пациентов с АНЦА-ИЗЛ занимает промежуточное положение между ИЛФ и ААВ без поражения легких [40]. На основании имеющихся данных в 2019 г. группой исследователей из клиники Мэйо (Рочестер, США) была предложена эмпирическая схема лечения МПО-АНЦА-ассоциированного ИЗЛ (табл. 1) [12].
| КТ-паттерн поражения легких | Клинико-лабораторная картина | Лечение |
|---|---|---|
| Примечание: ГКС – глюкокортикостероиды, ММФ – микофенолата мофетил, АЗА – азатиоприн | ||
| ОИП | Изолированное наличие МПО-АНЦА | Наблюдение + ежемесячное проведение общего анализа мочи для исключения гематурии |
| МПО-АНЦА + повышение уровня воспалительных маркеров | Наблюдение + ежемесячное проведение общего анализа мочи для исключения гематури | |
| МПО-АНЦА и МПА | Стандартная терапия МПА | |
| НСИП | Изолированное наличие МПО-АНЦА | ГКС + ММФ/АЗА |
| МПО-АНЦА + повышение уровня воспалительных маркеров | ГКС + ММФ/АЗА | |
| МПО-АНЦА и МПА | Стандартная терапия МПА | |
В будущем для лечения АНЦА-ИЗЛ могут быть использованы антифибротические лекарственные препараты, такие как пирфенидон и нинтеданиб, замедляющие прогрессирование и улучшающие выживаемость пациентов с ИЛФ. Данное предположение основано на том, что в 2019 г. завершено клиническое исследование, продемонстрировавшее эффективность антифибротической терапии у пациентов с фиброзирующими интерстициальными заболеваниями легких, не соответствующими ИЛФ, включая поражение легких в рамках системных заболеваний соединительной ткани [48]. В настоящее время Французской группой по изучению васкулитов проводится открытое исследование эффективности антифибротической терапии у пациентов с ААВ-ИЗЛ (NCT03385668) [24].
Поражение легких может вносить негативный вклад в долгосрочный прогноз пациентов с МПА. Так, в рядеисследований было выявлено повышение смертности в 2-4 раза в подгруппе пациентов ААВ-ИЗЛ по сравнению таковой у больных ААВ без поражения легких [24]. Средняя продолжительность жизни после установления диагноза ААВ-ИЗЛ составляет 3,5-6 лет при уровне 5-летней выживаемости 29-60%, что лишь незначительно превышает соответствующие значения при ИЛФ [36,37]. Основными причинами смерти при ААВ-ИЗЛ являются инфекционные осложнения, обострение ИЗЛ, а также прогрессирующее поражение легких с развитием терминальной дыхательной недостаточности [11].
В то же время в других исследованиях не было выявлено достоверных отличий между выживаемостью пациентов с ААВ и ААВ-ИЗЛ. Одной из вероятных причин этого может быть относительно короткий период наблюдения [24]. Вместе с тем, в когорте из 504 пациентов с ИЛФ 5- и 10-летняя смертность у АНЦАпозитивных пациентов была достоверно выше (61,3% и 85,7%), чем у АНЦА-негативных (37,6% и 70,5%). В данном исследовании, наличие ПР3-АНЦА (по сравнению с МПО-АНЦА), возраст более 65 лет, а также исходное значение DLCO менее 70% были ассоциированы с повышенной смертностью [25].
Заключение
ИЗЛ является одним из вариантов поражения легких при ААВ, оказывающим негативное влияние на течение и прогноз заболевания. У части пациентов ИЗЛ является первым проявлением болезни, развивающимся до появления других симптомов системного васкулита. Таким образом, исследование уровня АНЦА должно проводиться всем пациентам с идиопатическими интерстициальными пневмониями в рамках дифференциального диагноза.
Обсуждая подходы к терапии и тактику дальнейшего наблюдения, АНЦА-ИЗЛ можно разделить на две основные группы. К первой относятся интерстициальные поражения легких при наличии развернутой картины системного васкулита (как правило, МПА), требующего проведения стандартной иммуносупрессивной терапии и наблюдения ревматологами. Вторую составляют случаи ИИП, установленной в качестве первичного диагноза и сочетающейся с АНЦА. У таких пациентов важно оценить уровень воспалительных маркеров, а также провести активный поиск внелегочных, в том числе субклинических, проявлений ААВ, в частности, поражения почек, нервной системы, кожи, дыхательных путей, суставов. При наличии классификационных критериев ААВ необходимо проводить стандартную иммуносупрессивную терапию. Если данные в пользу ААВ отсутствуют, то назначение иммуносупрессивной терапии зависит от КТ-паттерна поражения легких. В части случаев обсуждается использование антифибротических препаратов.
Учитывая относительную редкость патологии, для получения достоверных данных об ее распространен ности в различных регионах, эффективности иммуносупрессивной и антифибротической терапии, определения оптимальной тактики ведения больных с АНЦА-ИЗЛ, требуется создание международных регистров пациентов и проведение проспективных рандомизированных клинических исследований. Кроме того, для сопоставления АНЦА-ИЗЛ с другими вариантами аутоиммунных поражений легких представляется обоснованным включение АНЦА в серологические критерии интерстициальной пневмонии с аутоиммунными признаками [49].
Идиопатические интерстициальные пневмонии
Рассмотрены интерстициальные заболеваний легких (ИЗЛ) с точки зрения классификации и терминологии, а также клинические формы и диагностические критерии идиопатических интерстициальных пневмоний (ИИП), идиопатического легочного фиброза и других ИЗЛ. Рассмо
Interstitial pulmonary diseases (IPD) were considered from the point of view of classification and terminology, and also clinical forms and diagnostic criteria of idiopathic interstitial pneumonia (IIP), idiopathic pulmonary fibrosis and other IPD. Approaches to the IIP treatment in patients were examined.
В группу интерстициальных заболеваний легких (ИЗЛ) включают несколько десятков отдельных нозологических форм, отличающихся по этиологии, особенностям патогенеза и морфологической картине, имеющих различную клинику и прогноз. Терминологические и классификационные подходы к этим заболеваниям неоднократно менялись, дополнительно затрудняя и без того непростую диагностическую работу с данной категорией пациентов. Даже сегодня, несмотря на наличие общепринятой классификации ИЗЛ, термин «интерстициальная пневмония» ассоциируется у врача скорее с вирусной инфекцией, чем с заболеванием, требующим обязательной морфологической верификации и имеющим довольно серьезный прогноз [1].
Напомним основные определения.
Пневмония — группа острых инфекционных (преимущественно бактериальных) заболеваний, характеризующихся очаговым поражением респираторных отделов легких с обязательным наличием внутриальвеолярной экссудации. Из рубрики «пневмония» исключены заболевания, вызванные физическими (лучевой пневмонит) или химическими («бензиновая» пневмония) факторами, а также имеющими аллергическое («эозинофильная пневмония») или сосудистое (инфаркт легкого вследствие ТЭЛА) происхождение. Воспалительные процессы в легких при кори, краснухе, гриппе и др. рассматриваются не в рубрике «пневмония», а в рамках соответствующих нозологических форм [2].
Пневмонит (альвеолит) — воспалительный процесс, часто иммунного, неинфекционного характера, затрагивающий преимущественно паренхиматозный интерстиций (альвеолярные стенки) и экстраальвеолярную соединительную ткань легких без обязательной внутриальвеолярной экссудации. Ряд авторов разграничивает понятия «пневмонит» и «альвеолит», предполагая, что при альвеолите воспалительный процесс локализуется преимущественно в альвеолах, а при пневмоните воспаление затрагивает и другие структуры паренхимы легких, однако практического значения такое разделение не имеет, и термины часто используются как синонимы [3].
Термин «пневмонит» отражает не конкретную нозологическую форму, а особенности патологического процесса. Поражение легких по типу пневмонита (альвеолита) может развиваться при самых разных заболеваниях: идиопатических интерстициальных пневмониях, лекарственных поражениях легких, системных заболеваниях соединительной ткани, гиперчувствительном пневмоните, саркоидозе и др. Следует учесть, что при каждом из этих заболеваний пневмонит является обязательным, но далеко не единственным проявлением поражения легких. Причины, клинические проявления, направления лечения и прогноз при этих заболеваниях различны, поэтому при выявлении признаков пневмонита такое большое значение имеют морфологическая верификация и дальнейшая нозологическая диагностика.
С современных позиций ИЗЛ представляют собой гетерогенную группу заболеваний, общими чертами которых являются поражение интерстициальной ткани легких по типу продуктивного пневмонита с последующим формированием фиброза, прогрессирующая одышка при нагрузке, непродуктивный кашель, крепитация, диффузные изменения при рентгенографии и компьютерной томографии легких, рестриктивные вентиляционные нарушения, снижение диффузионной способности легких и нарастающая дыхательная недостаточность [4].
В настоящее время в большинстве стран, в том числе и в России, используется классификация ИЗЛ, принятая Согласительной комиссии Американского торакального общества и Европейского респираторного общества (ATS/ERS, 2002 г.) [5]. Согласно этой классификации выделяют четыре группы ИЗЛ: ИЗЛ известной этиологии, гранулематозы, идиопатические интерстициальные пневмонии, другие ИЗЛ (рис.).
.gif)
Идиопатические интерстициальные пневмонии (ИИП) — типичные представители группы ИЗЛ неизвестной этиологии, имеющие много сходных клинических, рентгенологических и функциональных признаков, но принципиально разную морфологическую картину, обусловливающую особенности клиники, ответ на терапию и прогноз. Принципом построения клинико-патологической классификации ATS/ERS является соответствие каждой клинической форме ИИП определенного гистологического варианта ИИП (табл. 1).
Клинические особенности больных с различными видами ИИП приведены в табл. 2.
_575.gif)
Идиопатический легочный фиброз
Идиопатический легочный фиброз (ИЛФ) является одним из наиболее часто встречающихся заболеваний из группы ИИП. Синонимом ИЛФ являются «идиопатический фиброзирующий альвеолит» — термин, традиционно используемый в нашей стране.
Заболевание чаще всего встречается у пациентов в возрасте старше 50 лет. Основными жалобами больных являются нарастающая одышка и непродуктивный кашель. Начало болезни, как правило, незаметное, болезнь прогрессирует довольно медленно, пациенты успевают адаптироваться к своей одышке и на момент обращения имеют анамнез заболевания длительностью до 1–3 лет. Лихорадка и кровохарканье для больных ИФЛ не характерны. Другими симптомами могут быть общая слабость, артралгии, миалгии, изменение ногтевых фаланг в виде «барабанных палочек». Типичным аускультативным феноменом при ИЛФ является инспираторная крепитация, которую сравнивают с «треском целлофана». По мере прогрессирования заболевания появляются признаки дыхательной недостаточности и легочного сердца, снижение массы тела вплоть до кахексии. Данные лабораторного обследования неспецифичны. ИЛФ относится к рестриктивным легочным заболеваниям, поэтому характерными функциональными особенностями заболевания является снижение статических легочных объемов, выявляемое при бодиплетизмографии. Одним из ранних признаков заболевания является снижение DLCO. Спирометрический показатель FEV1/FVC находится в пределах нормы или повышен.
Наиболее частыми рентгенографическими признаками ИЛФ являются двусторонние изменения ретикулярного характера, более выраженные в нижних отделах легких. На ранних этапах развития заболевания может наблюдаться лишь некоторое уменьшение объема легочных полей и понижение прозрачности легких по типу «матового стекла». При прогрессировании заболевания ретикулярный паттерн становится более грубым, тяжистым, появляются округлые кистозные просветления, отражающие формирование «сотового легкого». Для уточнения рентгенологической картины целесообразно проведение мультиспиральной компьютерной томографии органов грудной клетки.
Поскольку возможности диагностики ИИП ограничены, а данные обследования не всегда специфичны, «золотым» диагностическим стандартом всех ИИП является биопсия легких: открытая либо торакоскопическая. Особая необходимость в выполнении биопсии возникает в случаях наличия не вполне типичной клинической и/или рентгенологической картины, возрасте пациента менее 50 лет, наличии системных признаков заболевании, быстром прогрессировании заболевания. Необходимым условием является преобладание пользы от постановки правильного диагноза над риском хирургической манипуляции.
Существует диагностический подход, позволяющий с большой вероятностью установить диагноз ИЛФ в тех случаях, когда проведении биопсии невозможно. Для этого необходимо, чтобы у пациента имелись четыре из четырех больших критериев и хотя бы три из четырех малых критериев.
Большие критерии
Малые критерии
Современная терапия ИЛФ построена, в основном, на противовоспалительной терапии (кортикостероиды и цитостатики (ЦС)), т. е. препаратах, способных воздействовать на воспалительные и иммунологические звенья развития заболевания. Базой такого подхода служит положение, что хроническое воспаление предшествует и неизбежно ведет к фиброзу и что агрессивное подавление воспаления может блокировать последующее формирование фиброзных изменений.
Широко используются три режима противовоспалительной терапии: монотерапия глюкокортикостероидами (ГКС), комбинация ГКС с азатиоприном и комбинация ГКС с Циклофосфаном. ATS/ERS рекомендует комбинированные режимы как более предпочтительные [6]. Терапия проводится, как минимум, в течение 6 месяцев. Обязательно тщательное мониторирование побочных эффектов терапии. При назначении цитостатиков мониторинг больных должен включать общий анализ крови еженедельно в течение первого месяца, затем один раз каждые 2–4 недели; при терапии Циклофосфаном требуется еженедельный анализ мочи на гематурию.
В случае выбора монотерапии ГКС начальная суточная доза преднизолона составляет 1 мг/кг идеального веса в сутки (максимум до 80 мг/сут). Через 4 недели проводится оценка переносимости такой терапии. Если произошло улучшение или стабилизация функциональных показателей, то в течение последующих 3 месяцев суточную дозу преднизолона уменьшают. При отсутствии ответа на стероиды добавляют азатиоприн [7].
Альтернативным подходом, сфокусированным на снижении избыточной депозиции матрикса в легких или ускорении распада коллагена, является антифибротическая терапия. К числу антифиброзных препаратов относятся D-пеницилламин, колхицин, интерферон гамма-1 b, пирфенидон.
Доказано повышение эффективности терапии при добавлении к противовоспалительным препаратам N-ацетилцистеина в дозе 600 мг 3 раза в сутки. В настоящее время ведущие эксперты при лечении ИЛФ отдают предпочтение схеме, включающей преднизолон, азатиоприн и N-ацетилцистеин [8].
Кроме медикаментозной терапии, как и при других заболеваниях легких, при развитии гипоксемии используется терапия кислородом. При развитии легочной гипертензии, кроме кислородотерапии, возможно использование вазодилататоров. Развитие инфекций трахеобронхиального дерева требуют использования антибактериальных и противогрибковых препаратов. Всем больным ИЛФ рекомендована регулярная вакцинация противогриппозными и антипневмококковыми вакцинами.
Другие идиопатические интерстициальные пневмонии (не-ИЛФ)
Неспецифическая интерстициальная пневмония (НИП) наряду с ИЛФ является одной из наиболее часто встречаемых форм ИИП. НИП может быть идиопатической, именно эта форма входит в группу ИИП. Однако морфологическая картина, соответствующая паттерну НИП, бывает и при поражении легких у больных с СЗСТ, гиперчувствительном пневмоните, радиационном пневмоните и т. д.
Клинические, лабораторные и функциональные показатели при НИП неспецифичны. Рентгенография грудной клетки чаще всего выявляет двусторонние изменения по типу «матового стекла» и ретикулярные изменения в нижних отделах легких.
Прогноз больных НИП более благоприятный, чем при ИЛФ. Клиническое течение и выживаемость больных зависят от выраженности легочного фиброза. Десятилетняя выживаемость при НИП составляет около 35%. Спонтанные случаи выздоровления без лечения при НИП неизвестны, терапия ГКС без или с добавлением цитостатиков приводит к улучшению или стабилизации приблизительно у 75% больных [9].
Криптогенная организующаяся пневмония
Синонимами криптогенной организующейся пневмонии (КОП) являются термины «облитерирующий бронхиолит с организующейся пневмонией» и «пролиферативный бронхиолит». КОП имеет четкие клинико-морфологические отличия от «изолированного» облитерирующего бронхиолита: наряду с поражением бронхиол наблюдается вовлечение в воспалительный процесс альвеол с наличием в их просвете организованного экссудата. КОП в большинстве случаев является идиопатическим, т. е. причина остается неустановленной. Среди установленных причин наибольше значение имеют СЗСТ (ревматоидный артрит и др.), осложнения лекарственной терапии (амиодарон, препараты золота и др.).
Заболевание чаще всего развивается у людей в возрасте 50–60 лет, мужчины и женщины болеют одинаково часто. КОП характеризуется острым или подострым течением, клиническая картина часто напоминает бактериальную пневмонию. Средняя продолжительность симптомов до момента постановки диагноза составляет 2–6 мес. Рутинные лабораторные тесты выявляют лейкоцитоз периферической крови (50%), повышение СОЭ и C-реактивного белка (70–80%).
Типичным рентгенологическим признаком КОП является наличие пятнистых, двусторонних (реже односторонних) плотных очагов консолидации субплевральной локализации. При КОП описана миграция легочных инфильтратов, чаще всего от нижних к верхним отделам. Дифференциальный диагноз КОП, кроме бактериальной пневмонии, проводят с хронической эозинофильной пневмонией, бронхоальвеолярным раком и лимфомой легких.
Спонтанное улучшение при КОП описано, но бывает редко. Терапия выбора при КОП — пероральные ГКС. Клиническое улучшение наступает уже через 1–3 суток от начала приема первой дозы, рентгенологические изменения обычно исчезают через несколько недель, общая длительность терапии ГКС составляет от 6 до 12 мес. При снижении дозы ГКС рецидивы заболевания возникают довольно часто, в такой ситуации вновь увеличивают дозу стероидов. Прогноз при КОП обычно благоприятный, большинство больных полностью излечивается при приеме ГКС. Однако в редких случаях наблюдается плохой ответ на стероиды и неуклонно прогрессирующее течение КОП. У таких больных рекомендовано использование цитостатиков [10].
Десквамативная интерстициальная пневмония
Десквамативная интерстициальная пневмония (ДИП) является довольно редким заболеванием из группы ИИП. Среди всех больных ДИП более 90% являлись курильщиками. Кроме того, описаны редкие случаи ДИП, ассоциированной с другими состояниями — СЗСТ, реакциями на лекарственные препараты, экспозицией к факторам внешней среды.
Клиническая картина заболевания типична для ИИП. Лабораторные, функциональные и рентгенологические показатели при ДИП не дают дополнительной информации.
При наличии сомнительной картины для исключения более агрессивных форм ИЗЛ рекомендовано проведение биопсии легких.
Отказ от курения является первым шагом лечения ДИП, так как показано, что данное мероприятие часто приводит к обратному развитию заболевания. Для большинства больных ДИП основным лечением является терапия преднизолоном в дозе 40–60 мг/сут. Начальная доза преднизолона обычно назначается на период 1–2 месяца, а затем дозу препарата постепенно снижают на протяжении 6–9 мес. На фоне терапии ГКС клиническое улучшение или стабилизация течения заболевания наблюдается приблизительно у двух третей больных ДИП. Значение цитостатиков при данной форме ИИП пока не ясно. 5- и 10-летняя выживаемость при ДИП составляет 95,2 и 69,6% соответственно [9].
Респираторный бронхиолит, ассоциированный с интерстициальным заболеванием легких
Респираторный бронхиолит, ассоциированный с интерстициальным заболеванием легких (РБ-ИЗЛ), — заболевание из группы ИИП, при котором респираторный бронхиолит сочетается с поражением альвеол и легочного интерстиция.
Данное заболевание встречается у курильщиков со стажем курения более 30 пачек/лет. Средний возраст больных колеблется от 30 до 40 лет. Клиническая картина и данные лабораторно-инструментального обследования типичны для ИИЛ.
Часто прекращение курения приводит к полному разрешению заболевания, в ряде случаев могут потребоваться небольшие дозы ГКС. Прогноз при РБ-ИЗЛ более благоприятный, чем при ИЛФ, но все-таки данное заболевание в ряде случаев может иметь неуклонно прогрессирующее течение и стать причиной смерти больных [11].
Лимфоцитарная интерстициальная пневмония
Лимфоцитарная интерстициальная пневмония (ЛИП) является одним из наиболее редко встречающихся заболеваний из группы ИИП. Как следует из названия, в основе заболевания лежит распространенная гомогенная лимфоцитарная инфильтрация легочного интерстиция. Морфологический диагноз ЛИП очень сложен, так как сходную гистологическую картину имеют некоторые заболевания, ассоциированные с массивной лимфоцитарной инфильтрацией ткани легких: псевдолимфома, первичная лимфома, лимфоматозный гранулематоз и др.
ЛИП встречается чаще всего у женщин, обычно в возрасте 40–60 лет. Большинство больных ЛИП — некурящие. Начало заболевания чаще всего незаметное, постепенное. Рентгенологическая картина ЛИП неспецифична.
Для постановки диагноза ЛИП во всех случаях требуется проведение открытой биопсии легких. Основу терапии ЛИП составляют ГКС. Дозы и длительность терапии приблизительно такие же, как и при других клеточных формах ИИП, например ДИП. На фоне противовоспалительной терапии улучшение или стабилизация заболевания отмечается у большинства больных (около 80%), хотя у небольшой группы их наблюдается медленное, но неуклонное прогрессирование заболевания. Кроме ГКС, у больных ЛИП применялись попытки терапии азатиоприном, циклофосфамидом, метотрексатом и циклоспорином [12].
Острая интерстициальная пневмония
Первые упоминания ОИП относятся к 1935 г., когда Hamman и Rich описали четырех больных с быстропрогрессирующей дыхательной недостаточностью, приведшей к смерти пациентов в течение 6 месяцев от начала болезни. На аутопсии был обнаружен выраженный распространенный фиброз легких [13]. Длительное время синдромом Хаммена–Рича назывались и заболевания с хроническим течением (в первую очередь ИЛФ), однако в настоящее время к синдрому Хаммена–Рича можно отнести только ОИП [14].
В современных руководствах ОИП рассматривается как заболевание, характеризующееся прогрессирующей дыхательной недостаточностью, приводящей в большинстве случаев к летальному исходу. Клиническая картина напоминает острый респираторный дистресс-синдром (ОРДС), однако при ОИП неизвестна причина заболевания и отсутствует вовлечение в процесс других систем организма (полиорганная недостаточность). В настоящее время в мировой литературе описано около 150 случаев ОИП, что связано не столько с редкостью заболевания, сколько со сложностью его диагностики [15].
Для ОИП характерно очень быстрое нарастание симптомов заболевания. Период от появления первых симптомов до обращения за медицинской помощью у большинства больных составляет не более 3 недель и очень редко превышает 2 месяца. Заболевание может развиться в любом возрасте и встречается одинаково часто у мужчин и женщин. Наиболее частыми симптомами ОИП являются непродуктивный кашель и диспноэ, лихорадка, миалгии, головная боль, слабость. При осмотре обращает на себя внимание тахипноэ, тахикардия, цианоз. При аускультации выслушивают крепитацию, реже — сухие свистящие хрипы.
Функциональные тесты неспецифичны и выявляют картину, характерную для других ИИП, однако полноценное функциональное исследование удается провести далеко не всегда. Характерным признаком ОИП является выраженная гипоксемия, часто рефрактерная к кислородотерапии, поэтому большинство больных, описанных в литературе, требовали проведения механической вентиляции легких.
Рентгенологическая картина при ОИП выявляет двусторонние пятнистые ретикулонодулярные тени, распространяющиеся практически на все легочные поля, за исключением реберно-диафрагмальных синусов, и плотные инфильтраты (консолидация). Типичными находками компьютерной томографии легких являются участки пониженной прозрачности паренхимы по типу «матового стекла», дилатация бронхов и нарушение легочной архитектоники. Изменения по типу «матового стекла» чаще всего имеют пятнистое распространение («географическая карта»).
Для морфологической верификации диагноза возможно проведение открытой или торакоскопической биопсии легких. Однако, к сожалению, из-за крайней тяжести больных с ОИП проведение данной диагностической процедуры чаще всего бывает невозможно. Все описанные в литературе морфологические изменения ОИП основаны на данных аутопсии или открытой биопсии легких, выполненной во время проведения больным ИВЛ.
Заболевание характеризуется фульминантным течением, прогноз плохой, летальность больных ОИП крайне высока и составляет, в среднем, 70% [16]. Дифференциальный диагноз ОИП чаще всего проводится с двусторонней бактериальной пневмонией или ОРДС. При ОРДС, как правило, известна причина (сепсис, травма, шок и т. д.); кроме того, ОРДС чаще всего бывает одной из составных частей полиорганной недостаточности.
Эффективной терапии ОИП в настоящее время не существует. Обязательными компонентами терапии ОИП являются кислородотерапия и респираторная поддержка.
Литература
М. В. Вершинина, кандидат медицинских наук, доцент
ГБОУ ВПО ОмГМА Минздравсоцразвития России, Омск