Синдром джоба что это
Синдром Чедиака — Хигаси — редкое заболевание, характеризующееся рецидивирующими пиогенными инфекциями, окулокутанным альбинизмом и наличием гигантских цитоплазматических гранул во многих клетках, преимущественно в лейкоцитах периферической крови. Болезнь наследуется аутосомно-рецессивно. Причину пиогенных инфекций связывают с нарушением бактерицидной активности лейкоцитов и нейтропенией.
Снижение бактерицидной активности зависит ют дефекта лизосом, а также от снижения способности лейкоцитов к хемотаксису. Кроме того, полагают, что фагоциты больных обладают повышенной активностью к аутофагоцитозу, что приводит к гиперспленизму и вторичной нейтропении. Дефект пигментации зависит от нарушений распределения меланофоров и аномального расположения меланосом.
У больных наблюдается гипопигментация кожи, радужной оболочки глаза и волос, повышенная чувствительность кожи к солнечному свету. Имеется, вероятно, связь нарушений пигментации с дефектом фагоцитарной системы. Течение инфекционных процессов также неблагоприятное, как и при ХГБ, наблюдаются они с периода новорожденности. Чаще, чем при ХГБ, поражается желудочно-кишечный тракт.
Диагноз синдрома подтверждается наличием гигантских включений в лейкоцитах, дающих положительную реакцию на пероксидазу.
Синдром Джоба характеризуется развитием рецидивирующих «холодных» абсцессов, вызванных золотистым стафилококком, встречается у девочек с нежной белой кожей и белокурыми или рыжими волосами. Некоторые считают его вариантом ХГБ. Предполагается аутосомно-рецессивная наследственность. А. Т. Devis и соавт., впервые подробно описавшие этог синдром в 1966 г., считали, что он является следствием дефекта местной резистентности ткани к стафилококку. Изучение киллерной способности лейкоцитов дает противоречивые результаты.
По одним данным, имеется дефект гексозомоносфатного шунта, как при ХГБ, по другим — киллерная способность лейкоцитов не нарушена [Quic P. О., Devis А. Т., 1973]. Абсцессы располагаются в коже, подкожной клетчатке, лимфатических узлах, легких, печени. Могут наблюдаться экзема, хронический ринит, отит. Содержание иммуноглобулинов нормальное, отмечается только повышение содержания IgE.
Абсцессы характеризуются отсутствием реактивных изменений со стороны окружающей ткани (отсюда их название «холодные» абсцессы), нет гиперемии, лейкоцитарного воспалительного вала. Данные биопсии кожи указывают на наличие вокруг абсцессов большого количества эозинофилов и базофилов при резком снижении количества нейтрофильных лейкоцитов.
Таким образом, у больных имеется отчетливое снижение резистентности к стафилококковой инфекции и нормальная способность к реакциям специфического клеточного и гуморального иммунитета, увеличение количества IgE и эозинофилия. Этиологическая и патогенетическая связь между всеми перечисленными изменениями неясна и требует дальнейшего изучения.
Дефект лейкоцитарного хемотаксиса Miller и соавт. в 1971 г., А. Соnstantopoules и соавт. в 1978 г. обнаружили у двух детей, у которых он сопровождался повторными тяжело текущими инфекциями и нейтропенией. Лейкоциты имели нормальную бактерицидную способность, но не были способны к хемотаксису in vitro. При этом количество лейкоцитов в костном мозге было нормальным, в периферической крови была стойкая нейтропения.
Окончатая проба с воспалением показывала появление миграции лейкоцитов только после инфузии свежей плазмы.
Наследственный детский агранулоцитоз (агранулоцитоз Костмана)—заболевание грудных детей, характеризующееся агранулоцитозом периферической крови и гипоплазией миелопоэза костного мозга. Впервые описан шведским педиатром Костманом в 1956 г. Характеризуется лихорадкой, кожными воспалительными поражениями и симптомами общего инфекционного заболевания. В периферической крови — нейтропения вплоть до полного исчезновения зернистых лейкоцитов. Прогноз неблагоприятный.
Этиология: аутосомно-рецессивная наследственность, возможно, сцепленная с Х-хромосомой, болеют мальчики. Заболевание описывается в семьях, где имеет место кровное родство родителей. Патогенез не установлен.
Синдром Джоба ( Синдром гипериммуноглобулина Е )
Синдром Джоба — это мультисистемное наследственное заболевание, в основе которого лежит первичный иммунодефицит. Возникает вследствие мутаций в генах STAT3, TYK2, DOCK8. Классическая триада симптомов включает экзематозный дерматит, рецидивирующие кожные инфекции, инфекционные поражения легких. Также отмечаются деформации черт лица, костные аномалии, нарушения формирования зубов. Для диагностики синдрома Джоба проводится иммунологическое исследование крови, генетическое тестирование. Пациентам назначается поддерживающее лечение, включающее антибиотики, антимикотики, противоаллергические средства.
МКБ-10

Общие сведения
Синдром Джоба (Иова) имеет второе название «гипериммуноглобулинемия Е», которое отражает основную патогенетическую особенность заболевания. Патология принадлежит к классу орфанных, в медицинской литературе имеются сведения о 250 случаях синдрома во всем мире. Впервые болезнь описана американским педиатром Дэвисом Старки в 1966 г., когда были установлены ее основные клинические признаки. Связь с гиперпродукцией IgE выявлена в 1972 г. врачом Ребеккой Баркли. Генетические предпосылки заболевания обнаружены в 2007 г.
Причины
Синдром Джоба имеет два типа наследования. При аутосомно-доминантном варианте болезнь вызвана мутацией в гене STAT3. В таком случае риск рождения больного ребенка составляет 50% независимо от пола, если один из родителей болен. Аутосомно-рецессивный вариант характеризуется дефектами генов TYK2 или DOCK8. Для развития патологии требуется, чтобы оба родителя были носителями мутантных аллелей, при этом вероятность наследования синдрома существует у 25% детей.
Патогенез
Ген STAT3 выполняет роль индуктора сигналов для нескольких видов интерлейкинов, которые участвуют в формировании иммунного ответа. Этот сигнальный путь контролирует активность провоспалительных и противовоспалительных молекул. При мутации гена нарушается дифференцировка Т-лимфоцитов Th-17, изменяется течение иммунологических реакций, в результате чего иммунитет неэффективно борется с микозами, бактериальными инфекциями.
Частые поражения кожи и легких при синдроме Джоба объясняются тем, что эти ткани наиболее сильно зависят от наличия Th-17, без которых они не могут реализовать свои местные антибактериальные механизмы. Как следствие, повышается восприимчивость к инфекциям, наблюдается их затяжное, рецидивирующее течение. Неиммунологические аспекты синдрома обусловлены нарушением ремоделирования тканей, сосудистыми патологиями.
Симптомы
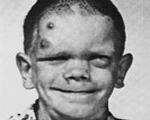
Клинические проявления синдрома Джоба возникают в младенческом возрасте. Сразу после рождения выявляются типичные аномалии строения черепа: у детей грубые черты лица, выступающие нос и подбородок, широкие крылья носа. Пор мере взросления становятся заметнее асимметрия лица, грубая кожа. Патологии опорно-двигательного аппарата дополняются гиперрастяжимостью связок, остеопенией, дегенеративными поражениями суставов.
Патогномоничный признак синдрома Джоба — множественные гнойничковые или экзематозные высыпания, возникающие в первые недели жизни. Сначала они локализованы на коже лица и волосистой части головы, позже распространяются по телу. Со временем сыпь трансформируется в экзематозный дерматит, ассоциированный с золотистым стафилококком. Специфично формирование холодных абсцессов, при которых отсутствует покраснение кожи, не повышается местная температура.
Гипериммуноглобулинемия Е также проявляется рецидивирующими инфекциями придаточных пазух носа, бронхов, легких. Их вызывают золотистые стафилококки, пневмококки, гемофильные палочки, а наибольшую опасность представляет инфицирование синегнойной палочкой. Пневмонии трудно поддаются лечению стандартными антибиотиками, вследствие постоянной иммуносупрессии возможно присоединение грибковой инфекции с развитием аспергиллеза легких.
Осложнения
Синдром Джоба отличается тяжелым течением, в патологический процесс вовлекаются все системы организма. Для аутосомно-доминантного типа заболевания характерны разрушение зубов, нарушения сроков смены зубного ряда, гипоплазия эмали. Поражения костно-мышечной системы представлены сколиозом, повышенной хрупкостью костей и риском переломов. Часто возникают сосудистые аномалии — аневризмы.
Аутосомно-рецессивный тип синдрома осложняется аллергическими расстройствами: поллинозом, атопическим дерматитом, бронхиальной астмой. В таких случаях четко прослеживается типичный «аллергический марш». Изредка появляются неврологические осложнения. Эта форма заболевания более опасна, поскольку больные имеют высокую вероятность развития лимфомы, рака кожи.
Основной причиной смертности пациентов с синдромом Джоба остаются тяжелые бактериальные легочные инфекции, которые чреваты сепсисом, септицемией, легочным кровотечением. Хронические пневмонии зачастую сопровождаются образованием пневматоцеле — воздушных кист, нарушающих процессы газообмена. Поражение бронхов завершается бронхоэктатической болезнью с частыми обострениями, которая также чревата генерализацией инфекции и смертельным исходом.
Диагностика
Обследованием больного занимается врач-неонатолог или педиатр. При внешнем осмотре обращают на себя внимание многочисленные высыпания, фурункулы, холодные абсцессы. Большое значение имеет выяснение семейной истории, определение степени риска болезни Джоба у ребенка. Для точной диагностики синдрома применяются следующие лабораторно-инструментальные методы:
Лечение синдрома Джоба
Консервативная терапия
Профилактическое лечение включает долгосрочный прием противостафилококковых антибиотиков (диклоксациллина, триметоприма-сульфаметоксазола), чтобы снизить риск кожных и респираторных инфекций. Для предупреждения микозов на фоне иммуносупрессии, усугубленной антибиотикотерапией, показаны противогрибковые препараты. С учетом клинической симптоматики используются:
Экспериментальное лечение
Трансплантация костного мозга рассматривается как перспективный вариант терапии, который снижает интенсивность клинических проявлений, повышает уровень резистентности организма. В литературе есть сообщения об успешном использовании моноклональных антител для нейтрализации избытка иммуноглобулинов Е в сыворотке крови. Ряд авторов говорят об эффективности плазмафереза в терапии синдрома Джоба.
Прогноз и профилактика
При адекватной комплексной терапии удается продлить срок и повысить качество жизни пациентов. Однако, несмотря на совершенствование протоколов лечения синдрома, большинство больных погибают в возрасте 20-40 лет от вторичных инфекционных осложнений. Первичная профилактика не разработана ввиду редкости заболевания. Вторичные превентивные меры включают раннюю диагностику, комплексную терапию, диспансерное наблюдение страдающих синдромом Джоба.
Синдром гипериммуноглобулинемии Е
Рубрика МКБ-10: D82.4
Содержание
Определение и общие сведения [ править ]
Синдром гипериммуноглобулинемии Е
Синонимы: синдром гипер-IgE и повторных инфекций (hyper immunoglobulin E recurrent infection syndrome), синдром Джоба (Job’s syndrome), синдром Бакли (Buckley syndrome), гипер-IgЕ-синдром, синдром Иова.
Синдром Джоба относится к редким; в мировой литературе описано немногим более 200 случаев. Синдром гипериммуноглобулинемии Е встречается с примерно одинаковой частотой во всех расово-этнических группах, с равной частотой среди мужчин и женщин.
Различают аутосомно-доминантный и аутосомно-рецессивный типы синдрома Джоба.
Аутосомно-доминантный тип преобладает, его диагностические критерии соответствуют типичным для «классического» синдрома гипер-IgE.
Аутосомно-рецессивный вариант встречается значительно реже. Характерны: повторные бактериальные инфекции, вызванные Staphylococcus aureus, Proteus mirabilis, Pseudomonas aeruginosa и Cryptococcus, упорная распространенная инфекция Molluscum contagiosum, рецидивирующий Herpes simplex и Varicella zoster, грибковые инфекции. Пневмонии при аутосомно-рецессивном варианте пневмоцеле не осложнялись.
Этиология и патогенез [ править ]
В основе аутосомно-доминантной (классической) формы гипер-IgE-синдрома лежат мутации в ДНК-связывающем или SH2-регионе гена STAT3 (3-го гена проводника сигнала и активатора транскрипции). Эти мутации не влияют на уровни белка STAT3 и совместимы с выживанием носителей подобных мутаций. У больных с аутосомно-рецессивной формой гипер-IgE-синдрома мутаций STATS-гена не обнаружено.
Характерны: эозинофилия в крови, мокроте, содержимом абсцессов; дефектный хемотаксис гранулоцитов; аномальное соотношение субпопуляций Т-лимфоцитов, дефектная продукция антител, сниженная продукция или ответ на ИЛ-4 и ИФНу. Однако ни один из этих дефектов не является характерным для всех больных с синдромом гипер-IgE. Чрезвычайно высокие уровни IgE (более 2000 КЕ/л) патогномоничны для этого состояния, но, по данным Grimbacher et al. (1999), 17% пациентов со временем теряли подобную степень гиперпродукции IgE, хотя не утрачивали характерной восприимчивости к инфекциям. Часть авторов выявляла снижение образования ИФНу и ФНО-α у больных с синдромом гипер-IgE, однако синтез ИЛ-2 и ИЛ-4 был нормальным.
Назначение ИФНу больным с синдромом гипер-IgE приводило у части больных к временному снижению продукции IgE, однако не влияло на клинические проявления заболевания.
Показано, что ИФНу и ИЛ-12 способны снижать образование IgE в культуре клеток больных с синдромом гипер-IgE in vitro, но не угнетать его полностью, тогда как нейтрализация ИЛ-4 или ИЛ-13, а также блокада ИЛ-6 или ФНО-α либо добавление в культуру клеток ИЛ-8 полностью блокировали продукцию IgE.
Сложная природа этого заболевания, одновременно поражающего кожу, кости, зубы, легкие, иммунитет и восприимчивость к инфекциям, указывает на некоторые нарушения в клетках или молекулярных процессах, общих для всех этих тканей.
Клинические проявления [ править ]
Наиболее частые проявления синдрома Джоба: экзема, абсцессы, пневмония, распространенный кандидоз, повышение IgE и эозинофилия. Рано появляющаяся (в первые дни или недели жизни) экзема упорного течения, нередко с атипичной областью высыпаний (зона разгибателей, спина, ягодицы, волосистая часть головы) встречается практически у всех больных.
У 83% пациентов отмечены аномалии лицевого скелета: асимметрия черепа, выдающийся лоб и умеренный прогнатизм, широкий крыловидный нос, широко расставленные глаза, утолщение ушных раковин, высокое небо.
Синдром гипериммуноглобулинемии Е: Диагностика [ править ]
Наиболее значимые диагностические критерии:
• чрезвычайно высокие уровни IgE в сыворотке (>1000 КЕ/л);
• рецидивирующие «холодные» абсцессы кожи и мягких тканей;
• повторные пневмонии с образованием бронхоэктазов и легочных булл;
• высокие уровни эозинофилов в периферической крови (более 700 клеток/мкл);
• рецидивирующие синуситы и отиты;
• задержка выпадения молочных зубов;
• характерные черты лица.
H.M. Chapel, S. Misbah, A.D.B. Webster предложили использовать определяющие, полезные и специфичные непостоянные диагностические признаки гипер-IgE-синдрома.
Диагноз вероятен, если имеются 3 или более определяющих признаков плюс 2 полезных критерия.
Дифференциальный диагноз [ править ]
Дифференциальный диагноз синдрома Джоба должен включать кистозный фиброз и хроническую гранулематозную болезнь, а также тяжелый атопический дерматит и ВИЧ-инфекцию. Также был описан клинически отличный аутосомно-рецессивный гипер-IgE-синдром.
Синдром гипериммуноглобулинемии Е: Лечение [ править ]
Специфического лечения гипер-IgE-синдрома не предложено.
Профилактика [ править ]
Прочее [ править ]
Гипер-IgE синдром
(Синдром гипериммуноглобулинемии Е; Синдром Джоба)
, MD, PhD, Cleveland Clinic Lerner College of Medicine at Case Western Reserve University
Аутосомно-доминантное: вызвано мутациями в гене STAT3 (преобразователь сигнала и активатор транскрипции гена 3)
Аутосомно-рецессивное: очевидно, вызвано гомозиготными нулевыми мутациями в генах TYK2 (тирозин-киназа 2) или DOCK8 (дедикатор цитокинеза 8)
Синдром гипериммуноглобулинемии IgE начинается в младенческом возрасте.
Симптомы и признаки синдрома гиперпродукции IgE
Гипер-IgE-синдром обычно проявляется в виде рецидивирующих стафилококковых абсцессов кожи, легких, суставов, внутренних органов; синусно-пульмональных инфекций; легочных пневматоцел; тяжелого зудящего эозинофильного дерматита.
У пациентов имеются грубые черты лица, нарушение прорезывания зубов, остеопения, рецидивирующие переломы. У всех пациентов отмечается эозинофилия тканей и крови при очень высоком уровне IgE ( > 2000 МЕ/мл 4800 мкг/л]).
Диагностика синдрома гиперпродукции IgE
Уровни IgE в сыворотке крови
Первичный диагноз синдрома гиперпродукции IgE основывается на симптомах и подтверждается измерением уровня сывороточного IgE.
Идентификация генных мутаций может проводиться с использованием генетического тестирования, которое осуществляют в основном для подтверждения диагноза или для прогнозирования моделей наследования.
Лечение гипер-IgE-синдрома
Профилактические антибиотики против стафилококковых инфекций
Иногда при тяжелой инфекции используют интерферон гамма
Лечение синдрома гиперпродукции IgE заключается в применении профилактической долгосрочной терапии противостафилококковыми антибиотиками (обычно триметоприм/сульфаметоксазол).
При дерматите кожу обрабатывают средствами для увлажнения, смягчающими кремами, антигистаминными препаратами. и, в случае подозрения инфекции, антибиотиками. При легочных инфекций применяют раннюю и агрессивную терапию антибиотиками.
В большинстве случаев при наличии остеопении необходимо проводить скрининг и лечиться в соответствии с действующими рекомендациями для пациентов без первичного нарушения иммунодефицита.
Интерферон гамма успешно используется для лечения инфекций, представляющих угрозу для жизни.
Была ли страница полезной?


Также интересно
Компания MSD и Справочники MSD
Компания «Мерк энд Ко. Инкорпорейтед», Кенилворт, Нью-Джерси, США (известная под названием MSD за пределами США и Канады) является мировым лидером в области здравоохранения и работает над оздоровлением мира. От разработки новых терапевтических методов для лечения и профилактики заболеваний — до помощи нуждающимся людям, мы стремимся к улучшению здоровья и благосостояния во всем мире. Справочник был впервые опубликован в 1899 году в качестве общественной инициативы. Теперь этот ценный ресурс приносит пользу в качестве руководства MSD за пределами Северной Америки. Узнайте больше о наших обязательствах в рамках программы Глобальная база медицинских знаний.