Синдром Геллера

Синдром Геллера – первазивное расстройство психического развития, характеризующееся внезапной утратой сформированных функций и навыков. Дебютирует в период с 2 до 10 лет. Ребенок утрачивает речь, способность решать интеллектуальные задачи, выполнять бытовые ритуалы. Не пытается использовать невербальные средства коммуникации, не интересуется играми. Становится раздражительным, тревожным, непослушным, гиперактивным. Диагностика проводится методом беседы, наблюдения, психологического исследования когнитивной сферы. Специфическое лечение не разработано, назначаются коррекционные занятия, симптоматическая медикаментозная терапия.

Общие сведения
Синдром впервые был описан австрийским педагогом Т. Геллером, в честь которого получил название. Синонимы – детское дезинтегративное расстройство, детская деменция, дезинтегративный психоз, симбиотический психоз, синдром Крамера-Польнова. Заболевание встречается редко, по различным данным, частота составляет 0,001-0,0017%. До недавнего времени считалось, что распространенность не зависит от пола, но развитие методов диагностики позволило различать случаи болезни Геллера с синдромом Ретта у девочек, в результате было установлено, что эпидемиологические показатели среди мальчиков выше в 4 раза. В МКБ-10 заболевание отнесено к рубрике «другие дезинтегративные расстройства детского возраста».
Причины синдрома Геллера
Этиологические факторы остаются неизвестными. Данные последних исследований указывают на связь патологического процесса с нейробиологическими механизмами центральной нервной системы. По результатам электроэнцефалографического обследования почти у 50% больных детей обнаруживается изменение электрической активности в головном мозге. Продолжается изучаться связь синдрома с судорогами, лейкодистрофией, болезнью Шильдера. Выдвигается предположение об инфекционном происхождении болезни, существовании фильтрующегося вируса – возбудителя малых размеров, недоступного исследованию под микроскопом.
Патогенез
Патогенетическая основа синдрома Геллера неизвестна, однако выделены закономерности развития патологических процессов. Заболеванию предшествует не менее двух и не более десяти лет нормального развития: ребенок частично или полностью овладевает речью, понимает обращения взрослых, использует социальные навыки. Внезапно возникают первые симптомы – гиперактивность, эмоциональные нарушения. В течение 6-12 месяцев распадается большинство приобретенных навыков, интеллектуальное развитие снижается до степени глубокой умственной отсталости (идиотии), теряется контроль опорожнения мочевого пузыря, кишечника. Затем регресс останавливается, состояние стабилизируется. Дальнейшее развитие, восстановление утраченных навыков происходит медленно при массивной психолого-педагогической помощи.
Симптомы синдрома Геллера
В продромальном периоде наблюдаются эмоциональные отклонения: ребенок проявляет своенравность, раздражительность, гневливость, тревожность. К аффективной вспыльчивости добавляется гиперактивность. Становятся недоступными сложные виды деятельности, требующие концентрации и распределения внимания, усидчивости, выполнения действий по образцу. Практически утрачиваются ранее существовавшие способности: сбор сложных моделей конструктора, рисование, разукрашивание, участие в сюжетно-ролевых играх («больница», «магазин»). Ребенок неусидчив, злится, отказывается от занятия при ошибках, затруднениях. На данном этапе признаки интеллектуального снижения отсутствуют, определить начало синдрома Геллера невозможно.
После нескольких месяцев эмоциональной неустойчивости, гиперактивности развиваются более специфические симптомы. Речь прогрессивно обедняется: сокращается словарный запас, развернутые фразы заменяются простыми, структурированные предложения – командами и односложными ответами («дай», «иди», «да», «нет»). В конечном итоге речь распадается, утрачивается разговорный язык, понимание обращений. Одновременно снижается интерес к сотрудничеству и общению. Ребенок замкнут, аутичен, не участвует в играх с другими людьми, не проявляет желания выполнять совместную деятельность. Исчезает стремление познавать окружающий мир, обедняются эмоции, упрощаются игровые навыки, развивается своеобразное «равнодушие».
Сложные двигательные навыки распадаются, заменяются стереотипными действиями. Больной неспособен выполнять ежедневные ритуалы – умываться, чистить зубы, надевать одежду, убирать игрушки, принимать пищу, ходить в туалет. Усиливаются проявления гиперактивности. Нарушается контроль мочеиспускания, опорожнения кишечника. Зачастую к признакам синдрома добавляются симптомы неврологической патологии. Спустя год после начала заболевания ребенок полностью утрачивает речевые, социальные, бытовые навыки.
Осложнения
После интенсивного прогрессирования болезни наступает стабильный период. Осложнения соматического и психического характера отсутствуют, но становится фактически невозможной социальная адаптация. При синдроме Геллера дети нуждаются в специальном обучении. Они не могут получать образование в средних и профессиональных учебных заведениях, не овладевают профессией, не создают семьи. Развитие медленное, поэтому им необходим постоянный посторонний уход, который в благоприятных случаях заменяется контролем. Заболевание ребенка изменяет социальное функционирование родителей, большинству приходиться отказываться от профессиональной деятельности, увлечений.
Диагностика
Диагностика синдрома Геллера зачастую начинается с консультации педиатра и невролога. Родители обращаются к специалистам на этапе постепенной утраты сформированных ранее навыков. Из-за редкости заболевания в первую очередь проводится осмотр ребенка и инструментальные обследования, позволяющие выявить более вероятные неврологические патологии – эпилепсию, опухоль и травму головного мозга. После исключения этих диагнозов ребенок направляется к врачу-психиатру. Специфическая диагностика включает:
Синдром Геллера дифференцируют с ранним детским аутизмом, болезнью Ретта, детской шизофренией. Основные диагностические критерии: период обычного развития, предшествующий болезни, не меньше 2 лет; появление симптомов до десятилетнего возраста; быстрый прогрессирующий распад навыков (от 6 до 12 мес.). В клинической картине преобладает дефицитарность, обеднение психических функций.
Лечение синдрома Геллера
Терапия детского дезинтегративного расстройства имеет общее направление с лечением раннего детского аутизма – основное внимание уделяется ранним и интенсивным мероприятиям, методы основаны на бихевиоральном походе, имеют высокую степень структурированности. Эффективность медикаментозного лечения не доказана, лекарства используются на первоначальном этапе для купирования выраженных поведенческих нарушений. Программа развития составляется индивидуально, к процессу реабилитации подключаются врачи, психологи, специальные педагоги, родители. В комплексный подход включены:
Прогноз и профилактика
Прогноз для детей, имеющих синдром Геллера, неблагоприятный, потерянные навыки остаются утраченными либо восстанавливаются крайне медленно, не полностью. При ранней интенсивной терапии около 20% пациентов приобретают способность изъясняться простыми фразами, осваивают самообслуживание, базовые бытовые и трудовые навыки, становятся социально активными в семье, реабилитационных и лечебных заведениях, которые посещают. Меры профилактики данного заболевания не разработаны, так как не установлены его причины и патогенетическая база. Исследования продолжаются.

Синдром Гурлера – это тяжелое наследственное метаболическое заболевание обмена веществ из группы мукополисахаридозов, характеризующееся избыточным накоплением гликозаминогликанов (ГАГ) в различных органах и тканях, что приводит к их выраженной дисфункции. Клиническая картина крайне разнообразна, включает задержку психомоторного развития, грубые деформации костей черепа и скелета, сердечно-легочные нарушения и пр. Диагностика основана на определении экскреции гликозаминогликанов с мочой и активности фермента альфа-идуронидазы в крови, данных молекулярно-генетических тестов. Лечение заключается в заместительной ферментной терапии.
МКБ-10

Общие сведения
Синдром Гурлера (болезнь Пфаундлера-Гурлер, мукополисахаридоз IH или первого типа) относится к лизосомным болезням накопления с аутосомно-рецессивным типом наследования, проявляется практически с первых месяцев жизни. Заболевание впервые было описано австрийским врачом-педиатром Гертрудой Гурлер. Патология считается одним из самых распространенных мукополисахаридозов, встречается повсеместно. Синдром Гурлера, по разным данным, выявляется у 1:40 000-1:100 000 новорожденных. Значимые гендерные статистические различия отсутствуют.
Причины синдрома Гурлера
Возникновение заболевания связано гетерогенными мутациями (мелкими делециями, дефектами сайта сплайсинга) гена IDUA, кодирующего синтез энзима альфа-L-идуронидазы. Ген локализован в коротком плече 4 хромосомы в локусе 4p16.3. Наиболее частые мутации гена – Q70X и W402X. Лизосомальный фермент α-L-идуронидаза регулирует метаболизм основных структурных компонентов межклеточного матрикса соединительной ткани – отвечает за деградацию гликозаминогликанов гепарансульфата и дерматансульфата.
Вследствие генетически детерминированного дефекта фермента происходит избыточное накопление ГАГ в лизосомах клеток. Наиболее значимым фактором риска развития синдрома Гурлера является наличие близкого родственника, страдающего этой болезнью. Если у одного из родителей имеется мутантный ген, вероятность рождения больного ребенка составляет 25%.
Патогенез
В результате нарушения внутрилизосомного гидролиза и последующего накопления межклеточных компонентов соединительной ткани высвобождаются воспалительные медиаторы – оксид азота, фактор некроза опухоли-альфа, развивается дисфункция органов. Поскольку соединительная ткань в той или иной степени входит в состав практически каждого органа, поражение носит мультисистемный характер.
Накопление мукополисахаридов в хрящевой ткани вызывает нарушение роста костей, их грубую деформацию. В стенках сосудов, клапанном аппарате сердца, мозговых оболочках развивается фиброз. При патологоанатомическом исследовании обнаруживается увеличение органов, гиперклеточность, дезорганизация. Отмечается уменьшение количества протеогликанов и коллагеновых волокон.
Симптомы
Клинические признаки заболевания начинают проявляться уже на первом месяце жизни. Иногда с рождения отмечается увеличение селезенки и печени, пупочные, паховые грыжи. Ярко выражены симптомы поражения опорно-двигательного аппарата. Достаточно быстро развиваются контрактуры и тугоподвижность суставов, формируются патологические изгибы позвоночника (кифоз поясничного отдела).
К концу первого года жизни лицо ребенка приобретает характерные черты по типу «гаргоилизма» – выступающие лобные бугры, широкая приплюснутая переносица, глазной гипертелоризм, толстые губы и язык, зауженная лицевая часть черепа. Типична задержка роста, который практически полностью останавливается к 4-5 годам. По мере прогрессирования заболевания присоединяются симптомы поражения центральной и периферической нервной системы.
Наблюдается заметное отставание психомоторного развития – интеллект снижен, речь неразвита. Дефектам речи также способствует формирующаяся нейросенсорная тугоухость. Из поведенческих расстройств отмечаются замкнутость и агрессия. Походка нарушена, тонус мышц снижен. Иногда возникают судороги вплоть до тонико-клонических пароксизмов.
Из других признаков синдрома Гурлера можно выделить частые инфекционные заболевания верхних дыхательных путей, рецидивирующие средние отиты, помутнение роговицы. Вследствие накопления ГАГ в миндалинах, трахее и надгортаннике постепенно сужается просвет дыхательной трубки, что повышает риск появления обструктивного апноэ сна.
Осложнения
Синдром Гурлера является тяжелым заболеванием с большим количеством осложнений. Основными причинами смерти считаются прогрессирующая хроническая сердечная недостаточность как исход формирующегося порока сердца и кардиомиопатии, дыхательная недостаточность из-за обструкции дыхательных путей, тяжелые инфекции (пневмония, менингит, туберкулез).
Гидроцефалия может привести к отеку головного мозга. Нарушения ритма сердца возникают вследствие фиброзного поражения миокарда. При компрессии спинного мозга возможны тетраплегия или нижняя параплегия, ухудшение или полная утрата тазовых функций. Иногда наблюдается потеря зрения и слуха.
Диагностика
Ввиду мультиорганного поражения пациентам с синдромом Гурлера необходим многопрофильный подход, поэтому курацию осуществляют врачи различных специальностей – педиатры, неврологи, кардиохирурги. Заподозрить наличие патологии помогают анамнестические данные и яркие фенотипические черты заболевания. Для подтверждения диагноза назначается дополнительное обследование, включающее:
Дифференциальный диагноз синдрома Гурлера проводится с другими наследственными метаболическими расстройствами – II, III типами мукополисахаридоза, ганглиозидозом, множественной сульфатазной недостаточностью. Поражение суставов следует отличать от неинфекционных артритов, ювенильного ревматоидного артрита.
Лечение синдрома Гурлера
Консервативная терапия
Пациенты подлежат обязательной госпитализации в стационар. Основным лекарственным патогенетическим лечением является пожизненная ферментная заместительная терапия (ФЗТ). Применяется рекомбинантная форма человеческой альфа-идуронидазы (ларонидаза). Введение этого препарата способствует восстановлению энзиматической активности, достаточной для гидролиза накопленных ГАГ и предотвращения их дальнейшего отложения.
Раннее использование ФЗТ позволяет замедлить прогрессирование неврологических расстройств и сердечной недостаточности, восстановить активные движения в суставах и позвоночнике, добиться регресса гепатоспленомегалии, исчезновения ночного апноэ. Также осуществляется следующая симптоматическая терапия:
Хирургическое лечение
Больным синдромом Гурлера в возрасте до 2 лет рекомендовано радикальное лечение, позволяющее избежать необходимости в пожизненной ферментотерапии, – трансплантация гемопоэтических стволовых клеток (ТГСК). Проводится пересадка костного мозга либо стволовых клеток пуповинной крови HLA-совместимых родственных доноров. ТГСК предотвращает развитие нарушений когнитивных функций. Предварительно назначается ФЗТ и иммуносупрессивная терапия.
При наличии соответствующих показаний выполняются следующие виды операций:
Реабилитация
Важной частью лечения являются реабилитационные мероприятия, включающие два основных аспекта. Массаж и лечебная физкультура необходимы для восстановления движений в суставах. Психолого-педагогическая помощь в виде систематических индивидуальных занятий направлена на развитие когнитивных функций, способствует более длительному сохранению интеллекта.
Прогноз и профилактика
Синдром Гурлера – тяжелое инвалидизирующее заболевание с высоким процентом летальности. Средняя продолжительность жизни больных без своевременно назначенного патогенетического лечения составляет 10 лет. Наиболее частыми причинами смерти выступают дыхательная и сердечная недостаточность, тяжелые бактериальные инфекции.
Единственной эффективной профилактикой развития патологии является прерывание беременности, если во время пренатальной диагностики была обнаружена низкая активность альфа-идуронидазы при исследовании ворсин хориона на 8-10 неделе беременности. При наличии близкого родственника, который страдает синдромом Гурлера, рекомендуется проведение ДНК-диагностики. Для предотвращения инфекционных осложнений назначается вакцинация от пневмококка, менингококка, гемофильной палочки.
Синдром Гулливера
Отсутствие понимания, где будет расположен реальный центр власти, по какой цепочке пойдут указания, обязательные для исполнения, а по какой – только рекомендованные (фигурально выражаясь: где расположится окошко выдачи зарплаты, преференций), возможно, является причиной того не всегда осознаваемого беспокойства, которое охватывает бюрократический класс, уже однажды обжегшийся на ложно понятой модернизации.
Действительно, когда Кремль одно время планировал обезопасить завоевания уходящего политического периода перестройкой президентской республики в частично парламентскую (чтобы не давать слишком много рычагов в руки непроверенному новому человеку), многие, например, слишком поторопились вступить в ряды «Справедливой России», ожидая найти в ней карьерные лифты. По какой-то причине Кремль снова переиграл ситуацию, решив не распылять власть (следовательно, и блага) в менее контролируемых массовых организациях, сосредоточившись исключительно на короткой рокировке между Кремлем и Белым домом.
Несомненно, что избирательный штаб Медведева уже понял опасность этого синдрома Гулливера в стране лилипутов, когда любое слово бог весть чем потом отзывается. О программе Медведева по большей части пока говорится, что она пишется, о реформе пенсионной системы – что она готовится, причем в обстановке глубочайшей секретности. И лучше бы для всех, чтобы так и оставалось. До тех пор, пока не наступит март и можно будет спокойно приступить к работе в новом качестве. А там уж и с портретами разберутся.
Синдром Гулливера: жизнь с iPhone SE после 6S Plus

Мне нравятся большие смартфоны. Два моих любимых устройства, которыми я постоянно пользуюсь в повседневной жизни — iPhone 6S Plus и Nexus 6P. Большие и классные: диагональ первого — 5,5 дюйма, второго — 5,7 дюйма. Настоящие «лопаты».
Я пишу с них тексты в метро. Смотрю фильмы и сериалы ночью, когда спит ребенок. Рассматриваю фотографии. Играю в игры. Мне нравится, как они держат заряд, как фотографируют и как выглядят.
И все это счастье в обмен на возможность управлять смартфоном одной рукой? Да запросто! 2016 — эпоха социальных сетей и цифрового контента, и гигантский экран — идеальный способ его потребления. Я в деле!
В общем — да, я люблю «лопаты». Но вот уже почти две недели живу без них. В моем кармане теперь iPhone SE, компактный смартфон Apple, с дизайном и размерами «пятерки» и монструозным железом 6S.
Кстати, где он? Мне снова кажется, что я его потерял. В сотый раз за эти четырнадцать дней.

iPhone SE ( apple.com)
На экране фотография: крохотный я с крохотным другом застыл на фоне крохотной… дальше с ходу не разглядишь. За несколько лет, проведенных в компании с гигантскими смартфонами, я успел забыть, что такое увеличение фотографий. Что ж, придется вспоминать. Как там говорил Стив Джобс? Pinch and Zoom?

iPhone SE ( apple.com)
А еще я практически перестал смотреть что-либо в метро, так что платная подписка на местный Wi-Fi («без рекламы! без регистрации! без перерывов на обед!») пропадает зря. Часто ловишь себя на мысли, что проще приехать домой и посмотреть все с экрана iPad, чем вглядываться в стеклышко SE.
Мне постоянно казалось, что я потерял телефон — чаще, чем за всю мою предыдущую жизнь. Ты совершенно не чувствуешь его веса, даже если он лежит в переднем кармане узких джинс. Перестав ощущать привычную тяжесть 6S Plus, я начинал шарить рукой в кармане куртки, где лежал SE в компании кредитных карт, не находил его и начинал паниковать. Еще один повод, перестать, наконец, носить пуховик — как-никак апрель на дворе.
Зажрался? Можно сказать и так, но я гарантирую: вам будет невероятно неудобно, перейди вы с пяти с половиной дюймов на четыре. Самое забавное, что работает эта история и в обратном направлении. Я назвал это «синдромом Гулливера»: когда ты пользуешься 6S Plus, iPhone SE кажется игрушечным, невероятно крохотным, и тебе сложно вообще представить, что это современный смартфон. Но на следующее утро та же «шестерка с плюсом» ощущается словно планшет, и так и норовит выпасть из рук (а ночью еще и упасть на лицо). Трансформация сознания от «боже, как можно пользоваться этой крохотулькой» до «в Apple что, думают, я Гаргантюа?» проходит часов за восемнадцать. Причем одновременно относиться хорошо к обоим смартфонам невозможно: либо «лопата», либо «лилипут», и никак иначе. Я, правда, в итоге со своим выбором определился…

Apple 6S Plus ( apple.com)
Как ни странно, главные достоинства SE вытекают из всего вышеперечисленного. Опустим ставший для меня откровением удивительный факт, что, оказывается, за поручень в автобусе можно держаться, не убирая смартфон в карман. Я ловил себя на мысли, что все реже и реже трачу время на интернет, меньше общаюсь в социальных сетях и «залипаю» на смешные картинки в сети по пути на работу или домой. iPhone SE — это замечательное устройство, фактически уникальное в своем роде. В компактном четырехдюймовом корпусе заключен один из самых мощных телефонов мира. Стильный, с узнаваемым классическим дизайном «пятерки». С сумасшедшей камерой. Быстрый, с прекрасной автономностью. Только вот использовал его я в основном как телефон: звонил, отправлял СМС, иногда отвечал на письма. А в остальное время смотрел не в экран, а вокруг. Если вы интернет-зависимый, и мечтаете вылечиться — SE может стать вашей нитью Ариадны из лабиринтов социальных сетей и мессенджеров.
Стоит ли его покупать? Если вы владелец 5S и не хотите переходить на «шестерки» из-за размеров — однозначно. Я не знаю, что еще могла бы сделать Apple, чтобы вам угодить. SE — это самый дешевый iPhone, к тому же лишенный каких-либо компромиссов. В ваш любимый смартфон завезли все самое лучшее, что есть у владельцев «старших» моделей, разве только 3D Touch нет. Да еще и в цвете «розовое золото».
А моя жизнь слишком сильно зависит от интернета, я запутался в мировой паутине, словно муха, да и не очень об этом жалею. Мне нужен большой экран и идущие с ним в комплекте большие возможности. Так что прямо сейчас я, пожалуй, встану из-за стола и вставлю сим-карту в потертый iPhone 6S.
Прости SE, но давай останемся друзьями.
Мукополисахаридозы – путь к диагнозу
Мукополисахаридозы (МПС) – это группа редких наследственных заболеваний, которые обусловлены дефицитом определенных лизосомных ферментов, участвующих в разрушении гликозаминогликанов (ГАГ), и характеризуются накоплением последних в различных органах и тканях. У части больных наблюдается медленное прогрессирование МПС, а типичные фенотипические признаки появляются в подростковом или зрелом возрасте, что в значительной степени затрудняется диагностику. Одним из типичных симптомов МПС является нарастающая тугоподвижность суставах, поэтому такие больные могут обращаться за помощью к ревматологам. Особенность поражения опорно-двигательного аппарата при МПС – отсутствие локальных и системных признаков воспаления. Важное диагностическое значение имеют системные проявления, такие как пупочная и паховая грыжа, изменение черт лица, помутнение роговицы, низкий рост, увеличение печени и селезенки, рецидивирующие инфекции дыхательных путей и средний отит и др. Для подтверждения диагноза определяют экскрецию ГАГ с мочой и активность лизосомных ферментов, а также проводят молекулярно-генетическое исследование.
Мукополисахаридозы (МПС) – это группа редких лизосомных болезней накопления, характеризующихся нарушением обмена гликозаминогликанов (ГАГ). Причиной каждого МПС является генетически обусловленный дефицит определенного лизосомного фермента, участвующего в разрушении ГАГ. Практически все МПС (за исключением МПС II) наследуются по аутосомно-рецессивному типу и с равной частотой встречаются у мальчиков и девочек. МПС II – это Х-сцепленное рецессивное заболевание, которое развивается у мальчиков, хотя описаны отдельные случаи и у девочек [1].
Накопление ГАГ в лизосомах различных тканей сопровождается разнообразными системными проявлениями, в том числе поражением опорно-двигательного аппарата, сердца, нервной системы, органа зрения и др., и приводит к прогрессирующему ухудшению функции внутренних органов. МПС в целом характеризуются тяжелым течением и неблагоприятным прогнозом, поэтому многие пациенты умирают в детском или подростковом возрасте. Однако возможно и более легкое течение заболевания, в частности МПС I и МПС VI, когда симптомы появляются в подростковом или старшем возрасте и нарастают более постепенно, а пациенты доживают до зрелого возраста [2,3]. В таких случаях диагноз нередко устанавливают с опозданием, а МПС длительно протекает под маской других болезней, прежде всего ревматических. Выделение легкого варианта течения МПС весьма условно, так как при медленном прогрессировании заболевания в конечном итоге развивается тяжелое поражение отдельных органов, которое приводит к инвалидизации пациентов и может потребовать оперативного вмешательства (например, протезирование тазобедренного сустава, имплантация искусственного клапана сердца, декомпрессия спинного мозга) [4]. МПС – это неоднородная группа заболеваний, которые имеют как общие фенотипические признаки, так и существенные различия (табл. 1, 2)
| • МПС I, II и VII – системные заболевания, поражающие различные органы и ткани, включая ЦНС; неврологические нарушения не бывают изолированными. |
| • МПС III характеризуется поражением ЦНС при отсутствии соматических проявлений. |
| • МПС IV поражает в основном опорно-двигательный аппарат и не сопровождается снижением интеллекта. |
| • При МПС VI наблюдается поражение различных органов и систем, интеллект остается нормальным. |
| Тип | Название | Фермент | Ген | Тип наследования | Лечение |
|---|---|---|---|---|---|
| МПС I | Синдромы Гурлера, Шейе или Гурлера-Шейе | α-L-идуронидаза | IDUA 4p16.3 | Аутосомно-рецессивный | Ларонидаза |
| МПС II | Синдром Хантера | Идуронат-2-сульфатаза | IDS Xq28 | X-сцепленный рецессивный | Идурсульфаза |
| МПС IIIA | Синдром Санфилиппо A | Гепаран-N-сульфатаза | SGSH 17q25.3 | Аутосомно-рецессивный | Разрабатывается |
| МПС IIIB | Синдром Санфилиппо В | α-N-ацетилглюкозаминидаза | NAGLU 17q21 | Аутосомно-рецессивный | |
| МПС IIIC | Синдром Санфилиппо С | Ацетил-КоА α-глюкозамин-ацетил-трансфераза | HGSNAT 8p11.1 | Аутосомно-рецессивный | |
| МПС IIID | Синдром Санфилиппо D | N-ацетилглюкозамин-6-сульфатаза | GNS 12q14 | Аутосомно-рецессивный | — |
| MПС IVA | Синдром Моркио А | Галактозамин-6 сульфатсульфатаза | GALNS 16q24.3 | Аутосомно-рецессивный | Элосульфаза |
| MПС IVB | Синдром Моркио В | β-Галактозидаза | GLB1 3p21.33 | Аутосомно-рецессивный | — |
| MПС VI | Синдром Марото-Лами | Арилсульфатаза B | ARSB 5q11.q13 | Аутосомно-рецессивный | Галсульфаза |
| МПС VII | Синдром Слая | β-Глюкуронидаза | GUSB 7q21.11 | Аутосомно-рецессивный | Разрабатыва |
| МПС IX | Синдром Натовича | Гиалуронидаза I | AH 3p21.3-p21.2 | Аутосомно-рецессивный | — |
Своевременная диагностика МПС сегодня приобрела особое значение, учитывая возможность заместительной терапии рекомбинантными ферментами, такими как идурсульфаза (МПС II), ларонидаза (МПС I), галсульфаза (МПС VI) и элосульфаза (МПС IVA), которые позволяют улучшить состояние больных или по крайней мере затормозить прогрессирование заболевания [5]. Ферментозаместительная терапия (ФЗТ) более эффективна, если ее начинают на более раннем этапе, когда еще отсутствуют необратимые проявления болезни.
Трудности диагностики МПС
МПС относятся к очень редким (орфанным) заболеваниям. В разных странах различные МПС регистрировали с частотой 1 на 16000-29000 живых новорожденных [6,7], а в 2007 году в Скандинавских странах распространенность МПС составила всего 4-7 случаев на 1 млн населения [8]. В связи с этим информированность врачей, особенно наблюдающих взрослых пациентов, о МПС низкая. Дополнительные сложности в диагностике возникают при более легком течении МПС, особенно при отсутствии типичных фенотипических проявлений, таких как низкий рост и характерные черты лица. Например, в зависимости от клинических проявлений и течения выделяют три формы МПС I – тяжелую (синдром Гурлера), промежуточную (синдром Гурлера-Шейе) и более легкую (синдром Шейе). Во всех случаях причиной заболевания является мутация гена, кодирующего α-L-идуронидазу. У пациентов с синдромом Гурлера симптомы появляются в раннем детском возрасте и часто наблюдается тяжелое поражение ЦНС, в то время как при синдроме Шейе симптомы менее выражены и возникают значительно позднее, а когнитивные расстройства обычно отсутствуют [9]. Два варианта течения заболевания – тяжелый и более легкий – возможны и при МПС VI (синдроме Марото–Лами), обусловленном мутациями гена, кодирующего арилсульфатазу В.
В клинике им. Е.М. Тареева за последние 3 года были обследованы 5 взрослых пациентов в возрасте от 20 до 33 лет с МПС VI. У трех из них диагноз был установлен в подростковом возрасте (от 7 до 16 лет), а у двух – в возрасте 23 и 30 лет, соответственно. Необ хо димо подчеркнуть, что хотя у двух последних пациенток наблюдалось замедленное прогрессирование заболевания, тем не менее, в обоих случаях на момент госпитализации в клинику имелось тяжелое поражение опорно-двигательного аппарата с резким ограничением подвижности в суставах, пороки клапанов сердца, стеноз шейного отдела позвоночника, нарушение проходимости дыхательных путей, поражение органа зрения и др. Обе пациентки были низкого роста (132 и 146 см) [4].
Замедленное прогрессирование течение иногда на блюдается и при МПС II (синдроме Хантера). Два года назад в нашу клинику был госпитализирован 42-летний пациент с МПС II, который был диагностирован в возрасте 13 лет на основании характерных изменений внешнего вида и наличия синдрома Хантера у старшего брата и подтвержден при энзимологическом (дефицит активности идуронат-2-сульфатазы) и молекулярногенетическом (мутация с.236С>А гена IDS в гемизиготном состоянии) исследованиях [10]. В течение длительного времени состояние пациента оставалось удовлетворительным. Успешно закончил школу, а затем институт. Работал инженером на заводе. С 30-летнего возраста прогрессирующее снижение чувствительности и боли в кистях и стопах, ухудшение зрения и выпадение центральных полей зрения, однако продолжал работать. Резкое ухудшение состояния, связанное с развитием сердечной недостаточности на фоне тяжелого порока аортального клапана, было отмечено только за год до госпитализации, т.е. в возрасте около 40 лет.
При обращении к ревматологу на МПС может указывать поражение суставов, не сопровождающееся признаками воспаления, такими как припухание, повышение СОЭ и/или уровня С-реактивного белка [12]. T. Rocha Siqueira и соавт. измеряли экскрецию ГАГ с мочой у 55 пациентов в возрасте от 3 до 21 года (в среднем 9 лет) с невоспалительной артропатией неясного генеза. У всех больных определялись дискомфорт или боль в суставах, а у 2/3 – скованность [12]. Экскре ция ГАГ была повышена у 1 из 55 больных. При дополнительном обследовании у 15-летней пациентки был установлен диагноз МПС II. Хотя очевидным ограничением этого исследования было небольшое число обследованных пациентов, тем не менее, полученные данные указывают на возможную роль скрининга в диагностике более легких форм МПС.
Как заподозрить МПС?
В настоящее время известно 11 лизосомных ферментов, дефицит которых приводит к развитию 7 типов МПС [13]. Замедленное прогрессирование заболевания и более поздняя диагностика чаще отмечаются у пациентов с МПС I, IV, VI и VII, в то время как другие типы МПС обычно характеризуются тяжелым течением и более короткой продолжительностью жизни. Следует отметить, что в задачи практического врача не входит дифференциальная диагностика различных МПС – вполне достаточно заподозрить этот диагноз и направить пациента на консультацию к генетику и/или провести скрининговое исследование (определение экскреции ГАГ с мочой).
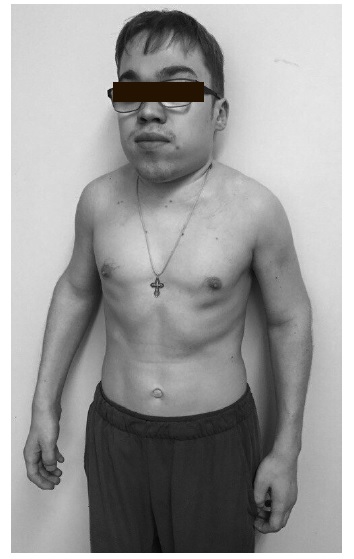 Рис. 1. Два брата с МПС II. Низкий рост, короткая шея, характерные черты лица, ограничение подвижности в плечевых суставах, пупочная грыжа
Рис. 1. Два брата с МПС II. Низкий рост, короткая шея, характерные черты лица, ограничение подвижности в плечевых суставах, пупочная грыжа
Хотя МПС представляют собой неоднородную группу болезней и отличаются по тяжести течения и частоте поражения центральной нервной системы, тем не менее, в целом клинические проявления некоторых из них достаточно однотипны и позволяют предположить наличие заболевания, особенно у пациентов старшего возраста при наличии типичного фенотипа. При осмотре пациентов с МПС прежде всего обращают на себя внимание низкий рост, непропорциональное строение скелета (короткие туловище и шея, длинные конечности), а также грубые черты лица, толстые губы, увеличение языка, запавшее переносье, увеличение расстояния между глазами (гипертелоризм) (рис. 1, 2). При тяжелом течении МПС рост пациентов не превышает 95-100 см, хотя при медленном развитии заболевания может достигать 140-150 см. Например, в нашей серии наблюдений рост 5 взрослых пациентов с МПС VI варьировался от 132 до 153 см, а рост 42-летнего пациента с МПС II составлял 158 см. В крупном исследовании среди 121 пациента с МПС VI доля взрослых составляла около 25% [14]. Средний рост больных в возрасте 19-24 и 25-56 лет равнялся 142,7±20,1 и 157,0±8,5 см, соответственно. Таким образом, по крайней мере у части больных МПС рост может быть фактически нормальным.
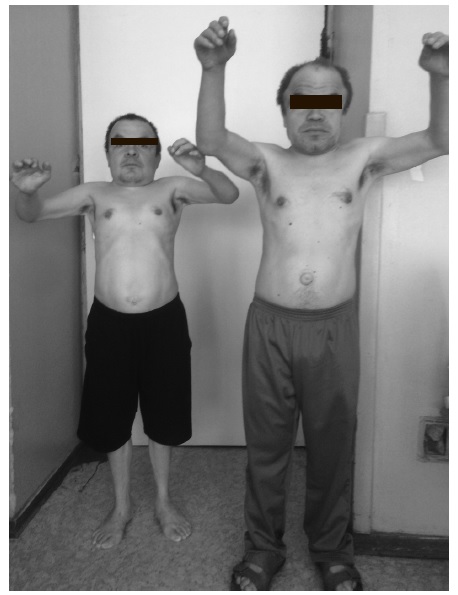 Рис. 2. ПациентсМПСVI
Рис. 2. ПациентсМПСVI
При всех МПС развивается тяжелое поражение опорно-двигательного аппарата (множественный дизостоз), которое проявляется тугоподвижностью и контрактурами суставов (в большей степени ухудшается разгибание), деформацией кистей (“когтистая лапа”) (рис. 3) и позвоночника (кифоз, сколиоз), воронкообразной грудной клеткой. Наблю даются недоразвитие таза, дисплазия головок бедренных костей и вальгусное положение шейки бедренной кости. Ограничение подвижности суставов отмечается уже в детском или подростковом возрасте, постепенно нарастает и в конечном итоге служит причиной инвалидизации больных.
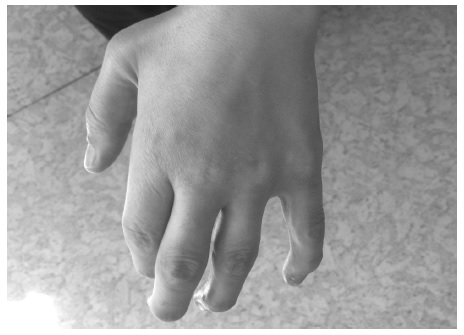 Рис. 3. Характерныеизменениякистиупациента с МПС III
Рис. 3. Характерныеизменениякистиупациента с МПС III
Для МПС IV (синдрома Моркио), в отличие от других типов МПС, типично развитие гипермобильности суставов, обусловленной деформацией метафизов, гипоплазией костей и деградацией соединительной ткани, окружающей суставы [15].
У пациентов с МПС часто наблюдаются обструкция глотки, верхних и нижних дыхательных путей, связанная с увеличением языка и миндалин, сужением трахеи, утолщением надгортанника и голосовых связок, отложением ГАГ в слизистой оболочке бронхов. Обструкция дыхательных путей сопровождается затрудненным дыханием и громким храпом с эпизодами апноэ во время сна. Характерно развитие рецидивирующего среднего отита, вызывающего прогрессирующую тухоугость, которая обусловлена как кондуктивными, так и нейросенсорными механизмами. Причинами нарушения функции дыхания могут быть также небольшие размеры и малоподвижность грудной клетки, растя жение живота в сочетании с кифозом, сколиозом и значительным поясничным лордозом, а также рецидивирующие инфекции нижних дыхательных путей.
Еще одно типичное проявление МПС – поражение клапанов сердца, частота которого достигает 60-90%. С. Wippermann и соавт. обследовали 84 больных в возрасте от 1 до 47 лет с различными типами МПС [16]. Частота недостаточности митрального и/или аортального клапана составила 75,0%, однако тяжелая митральная или аортальная регургитация наблюдалась только в 4,8% и 8,3% случаев, соответственно. Частота пороков клапанов сердца достигала 89-100% у больных МПС I, II и VI, но была ниже у пациентов с МПС III и IV – 3366%. В другом исследовании у 28 больных МПС VI частота поражения митрального клапана составила 96%, трикуспидального – 71% и аортального – 43% [17]. Следует отметить, что, в отличие от некоторых других лизосомных болезней накопления, таких как болезнь Фабри, для МПС не характерно тяжелое поражение миокарда.
У большинства больных МПС I, VI и VII часто отмечается помутнение роговицы, в то время как при других типах МПС оно отсутствует [18].
У пациентов с тяжелыми формами МПС I и II наб лю дается поражение ЦНС (поведенческие расстрои ̆ства, задержка умственного развития, ухудшение интеллекта, тяжелая когнитивная дисфункция) [10]. Выраженные неврологические и когнитивные расстройства характерны также для МПС III. В то же время у большинства пациентов с МПС VI сохраняется нормальный интеллект.
МПС I, II и VI могут привести к развитию синдрома запястного канала, проявляющегося стойкой болью и онемением пальцев кисти в результате сдавления срединного нерва между костями и сухожилиями мышц запястья. Возможно также сдавление спинного мозга вследствие сужения спинно-мозгового канала и нестабильности атлантоаксиального канала. Компрессион ная миелопатия может осложниться слабостью в нижних конечностях и спастической параплегией или квадриплегией.
Диагноз и дифференциальный диагноз
Алгоритм диагностики и дифференциальной диагностики МПС у пациентов с поражением опорно-двигательного аппарата представлен на рис. 4 [19]. Если контрактуры суставов определяются у новорожденного ребенка, то наиболее вероятен диагноз артрогрипоза – заболевания, характеризующегося врожденными контрактурами двух и более суставов несмежных областей в сочетании с мышечной гипо- или атрофией. Различают артрогрипоз с поражением верхних и/или нижних конечностей, генерализованный и дистальный варианты. Артрогрипоз – это не самостоятельная нозологическая форма, а скорее физический симптом, который может быть обусловлен различными причинами, например, ограничением движений плода во время его развития (многоводие, маловодие, пороки развития и опухоли матки, многоплодная беременность), нарушением развития мышц (вирусные инфекции), генетическими факторами и др.
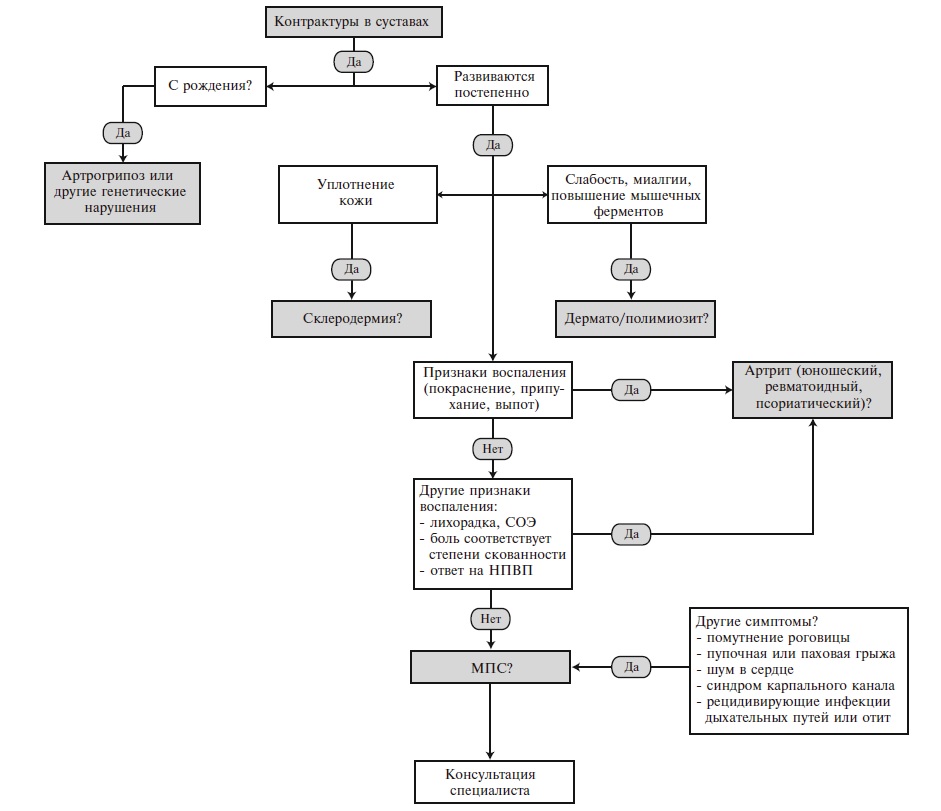 Рис. 4. Алгоритм дифференциальной диагностики у пациентов с тугоподвижностью/контрактурами суставов
Рис. 4. Алгоритм дифференциальной диагностики у пациентов с тугоподвижностью/контрактурами суставов
Боли и тугоподвижность суставов, появляющиеся в детском или подростковом возрасте, могут имитировать ревматические заболевания, в частности юношеский идиопатический артрит, ревматоидный или псориатический артрит. Основное значение для дифференциальной диагностики с этими заболеваниями имеют отсутствие воспалительной боли (т.е. боли, возникающей по утрам и сопровождающейся скованностью, которая уменьшается на фоне физической активности), локальных (припухание и болезненность при пальпации суставов) и системных (повышение температуры тела и/или СОЭ и уровня С-реактивного белка) признаков воспаления (рис. 4) [19]. Глюкокортикостероиды неэффективны, хотя нестероидные противовоспалительные препараты могут несколько уменьшить имеющиеся симптомы.
Признаки поражения суставов, появляющиеся в более старшем возрасте, часто расценивают как первичный остеоартроз. Дифференцировать поражение опорно-двигательного аппарата при МПС с этим забо леванием позволяют развитие артропатии в подростковом или молодом возрасте при отсутствии факторов риска первичного остеоартроза и наличие других типичных проявлений генетического заболевания (карликовый рост, измененные черты лица, порок клапана сердца, помутнение роговицы и т.п.) (табл. 3).
| Поражение опорно-двигательного аппарата |
| Контрактуры суставов, развивающиеся в раннем возрасте и не сопровождающиеся признаками воспаления или эрозивными изменениями костей |
| “Когтистая лапа” |
| Деформация позвоночника (сколиоз, кифоз, лордоз) |
| Рентгенологические признаки множественного дизостоза |
| Другие клинические проявления |
| Нарастающая “грубость” черт лица |
| Помутнение роговицы |
| Короткая ригидная шея |
| Частые респираторные инфекции, рецидивирующий средний отит, заложенность носа, шумное дыхание/храп |
| Шум в сердце |
| Пупочные и/или паховые грыжи |
| Низкий рост |
| Нарушение походки |
| Увеличение живота за счет печени и селезенки |
Скрининговым методом диагностики МПС является измерение экскреции ГАГ с мочой. Определение типа ГАГ в моче (дерматансульфат, гепарансульфат, хондороитинсульфат и кератансульфат) с помощью тонкослойной хроматографии или электрофореза имеет определенное значение для дифференциальной диагностики МПС, однако результаты этих исследований все же не позволяют установить окончательный диагноз. Экскреция ГАГ с мочой у детей, подростков и молодых людей с МПС обычно превышает таковую у здоровых людей сопоставимого возраста [13]. Однако у взрослых людей с МПС, особенно с более легкими и медленно прогрессирующими формами заболевания, она может оказаться близкой к норме. Соответственно, следует осторожно интерпретировать результаты этих тестов и продолжать обследование, если диагноз МПС представляется вероятным на основании клинических данных.
Следующий этап диагностики – определение активности лизосомных ферментов в высушенных пятнах крови, лейкоцитах или фибробластах. Анализ высушенных пятен крови обычно проводят в тех случаях, когда образец необходимо отправить в лабораторию, находящуюся в другом городе или стране. Более надежным считают исследование лейкоцитов, выделенных из цельной крови, или культивированных фибробластов.
Для подтверждения диагноза проводят молекулярногенетическое исследование, которое необходимо также для выявления носителей мутантных генов и пренатальной диагностики.
Лечение мукополисахаридозов
Для патогенетической терапии МПС применяют рекомбинантные формы ферментов, дефицит которых лежит в основе развития соответствующего заболевания, в том числе ларонидазу для лечения МПС I, идурсульфазу – МПС II, галсульфазу – МПС VI, элосульфазу альфа – МПС IVa (в Российской Федерации последний препарат не зарегистрирован). Все препараты предназначены для внутривенного введения. Их эффективность и безопасность установлены как в рандомизированных, двойных слепых, плацебо-контролируемых исследованиях, так и длительных наб людательных исследованиях, позволивших изучить отдаленные эффекы ФЗТ [20].
Эффективность и безопасность галсульфазы оценивали в рандомизированном, двойном слепом, плацебоконтролируемом, 24-недельном исследовании 3 фазы у 39 больных МПС VI [21]. Критериями эффективности были толерантность к физической нагрузке и экскреция ГАГ с мочой. Лечение галсульфазой в течение 24 недель по сравнению с плацебо привело к значительному увеличению пройденной за 12 минут дистанции (р=0,025) и скорости подъема по лестнице (р=0,053) и достоверному снижению экскреции ГАГ с мочой (p
Заключение
МПС – это группа редких заболеваний, которые обычно диагностируют поздно вследствие низкой информированности врачей о лизосомных болезнях накопления. Наибольшие диагностические трудности возникают при более легких формах МПС, которые характеризуются медленным развитием соматических проявлений и стертостью типичных внешних признаков. Выделение легких, или ослабленных (attenuated), форм МПС весьма условно, так как в конечном итоге у таких больных развиваются инвалидизирующие осложения, часто требующие оперативного лечения. Одним из типичных симптомов МПС I, II и VI является нарастающая тугоподвижность в суставах, поэтому такие больные могут обращаться за помощью к ревматологам. Особенность поражения опорно-двигательного аппарата при МПС – отсутствие локальных (припухания суставов и болезненности при их пальпации) и системных (повышения температуры тела и/или СОЭ и уровня С-реактивного белка) признаков воспаления. Исключить остеоартроз позволяют молодой возраст пациента и отсутствие типичных факторов риска дегенеративных заболеваний суставов. Важное диагностическое значение имеют системные проявления, такие как пупочная и паховая грыжа, изменение черт лица, помутнение роговицы, низкий рост, увеличение печени и селезенки, рецидивирующие инфекции дыхательных путей и средний отит и др. Если заподозрен диагноз МПС, то необходимо определить экскрецию ГАГ с мочой, а также измерить активность лизосомных ферментов и провести молекулярно-генетическое исследование для подтверждения диагноза.