Пфайффера синдром
OMIM 101600
Наша команда профессионалов ответит на ваши вопросы
Выделяют три типа ACS5. Классический ACS5 относится к типу 1. Тип 2 характеризуется черепом в форме клеверного листа и руками и ногами Пфайффера, а также анкилозом локтей. Тип 3 похож на тип 2, но у больных череп обычной формы. У больных отмечается тяжелый глазной проптоз, короткое переднее основание черепа. Также для типа 3 характерны различные лицевые изменения. Раннее отставание в развитии отмечается при типах 2 и 3. Эти типы описаны только как спорадические случаи.
В ряде семей было обнаружено сцепление ACS5 с 8р11.2-р11.1, где расположен ген рецептора фактора роста фибробластов-1 (FGFR1, MIM 136350). В нем найдена мутация p. 252 Pro>Arg, приводящая к ACS5. Описано несколько семей с характерными изменениями ног. Изменения черепа могут присутствовать, однако их может и не быть. Большой палец ног широкий и плоский, он обычно отклонен в сторону, также отмечается синдактилия второго и третьего пальцев. Предполагают, что при этих характерных изменениях ног даже в изолированных случаях без краниосиностоза надо проводить поиск мутации p. 252 Pro>Arg в экзоне 7a гена FGFR1.
Кроме того мутации в гене рецептора фактора роста фибробластов-2 FGFR2 (fibroblast growth factor receptor 2; MIM 176943) приводят к ACS5. Ген FGFR2 локализован на хромосоме 10q26 и состоит из 20 экзонов. Известно 26 мутаций, в основном располагающихся в экзонах 7 и 9 гена, приводящих к развитию заболевания. Однако описаны семьи, в которых отсутствует сцепление как с локусом FGFR1 на хромосоме 8, так и с локусом FGFR2 на хромосоме 10. Ряд мутаций в гене FGFR2, приводящих к ACS5, встречаются при синдроме Крузона (MIM 123500). Мутация р. 290 Trp>Cys описана при ACS5. Но в кодоне 290 описаны мутации p.290 Trp>Arg, приводящая к классической форме синдрома Крузона, и p.290 Trp>Gly, выявленная у больного атипически мягкой формой синдрома Крузона. Т.о. кодон 290 может быть основной «горячей точкой» в гене FGFR2. Другой «горячей точкой» является цистеин в положении 342. Эти два кодона локализованы в экзонах 7 и 9, кодирующих иммуноглобулинподобный домен рецептора. Отмечается эффект возраста отца при образовании мутации de novo. При исследовании происхождения мутаций de novo с помощью внутригенных полиморфных маркеров во всех информативных случаях показано их отцовское происхождение.
В Центре Молекулярной генетики проводится поиск мутаций в экзонах 7 и 9 гена FGFR2 и экзоне 7a гена FGFR1 методом прямого автоматического секвенирования.
Синдром Пфайффера
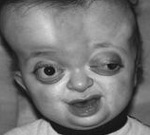
Синдром Пфайффера – генетическое заболевание с аутосомно-доминантным механизмом наследования, характеризующееся нарушением формирования костей черепа и конечностей. Симптомами являются деформации черепа (в результате краниосиностоза), костей пальцев рук и ног. Некоторые формы заболевания характеризуются глухотой, нарушением интеллектуального развития, стоматологическими патологиями. Диагностика синдрома Пфайффера производится на основании данных настоящего статуса больного, рентгенологических исследований, молекулярно-генетических анализов. Специфическое лечение отсутствует, применяют паллиативные мероприятия для предотвращения тяжелых осложнений (в том числе и хирургического характера) и симптоматическую терапию.
Общие сведения
Синдром Пфайффера – врожденное генетически обусловленное нарушение формирования черепа и конечностей, которое характеризуется краниосиностозом, деформацией лица, расширением и укорочением пальцев, частыми синдактилиями. Впервые это заболевание было описано в 1964 году немецким педиатром Рудольфом Пфайффером, в дальнейшем было выявлено несколько клинических форм патологии.
По данным современной генетики, синдром Пфайффера является аутосомно-доминантным состоянием с полной пенетрантностью, половое распределение больных не имеет особенностей – с одинаковой частотой болеют как мальчики, так и девочки. Встречаемость точно не определена, по некоторым данным она составляет примерно 1:100 000 новорожденных. Каких-либо национальных или расовых особенностей в распределении этого заболевания также не выявлено. В отношении синдрома Пфайффера крайне важна ранняя диагностика, так как от времени начала паллиативного лечения во многом зависит прогноз заболевания и качество дальнейшей жизни больного.
Причины синдрома Пфайффера
Несмотря на то, что синдром Пфайффера является наследственным заболеванием с аутосомно-доминантным механизмом передачи, большинство случаев этой патологии являются следствием спонтанных герминативных мутаций в половых клетках родителей. При этом за развитие состояния отвечают дефекты двух генов, кодируемые ими белки обладают сходными функциями.
По причине того, что вышеуказанные белки обладают очень похожими функциями, патогенез синдрома Пфайффера при дефектах гена FGFR1 и FGFR2 тоже весьма схож. В результате миссенс-мутаций изменяется структура экспрессируемого белка, из-за чего полученный рецептор становится неспособен полноценно связываться с веществом-стимулятором (фактором роста фибробластов) и передавать информацию внутрь клетки. Так как фибробласты активно участвуют в процессах остеогенеза в эмбриональный период и в первые годы жизни ребенка, такое изменение рецепторов ведет к разнообразным костным патологиям.
Доказательством этого является тот факт, что дефекты гена FGFR1, помимо синдрома Пфайффера, приводят к тригоноцефалии 1-го типа и ряду других нарушений, а FGFR2 – к неспецифическому краниосиностозу, синдрому Крузона и иным похожим патологиям. При синдроме Пфайффера происходит преждевременное сращение костей черепа, что становится причиной деформации головы и лица, повышения внутричерепного давления – это, в свою очередь, способно вызывать ряд вторичных нарушений от нейросенсорных расстройств до глубокой умственной отсталости.
Классификация и симптомы синдрома Пфайффера
На сегодняшний день выделяют как минимум три клинические формы синдрома Пфайффера, которые различаются между собой выраженностью нарушений, течением и прогнозом заболевания. Причина такого различия лежит в молекулярно-генетических механизмах развития патологии – 1-й тип обусловлен единственной мутацией гена FGFR1 (252Pro-Arg) и некоторыми нарушениями структуры FGFR2, тогда как 2-й и 3-й типы вызваны различными вариантами дефектов только гена FGFR2.
Более тонкие особенности патогенеза каждой формы заболевания в настоящее время находятся в стадии изучения. Благодаря характерной клинической картине, специалисты могут определить разновидность синдрома Пфайффера, основываясь только на симптомах патологии. Молекулярно-генетический анализ во многих случаях необходим лишь для окончательного подтверждения диагноза.
Синдром Пфайффера 1-го типа является наиболее распространенным и благоприятным в прогностическом отношении вариантом заболевания. При этом состоянии уже с самого рождения ребенка отмечаются аномалии развития лица – гипертелоризм, недоразвитие верхнечелюстных костей, расширенные плоские переносица и спинка носа. Иногда могут отмечаться расщепление твердого нёба и экзофтальм. При этой форме синдрома Пфайффера характерные изменения формы черепа сводятся к увеличению его высоты и длины, очень редко отмечается асимметрия. В дальнейшем могут возникать аномалии зубов – искривление зубного ряда, гипоплазия, склонность к кариесу. Деформации костей конечностей сводятся к расширению фаланг первых пальцев на руках и ногах, может отмечаться частичная синдактилия.
Синдром Пфайффера 2-го типа – более тяжелая форма заболевания, проявляющаяся значительной выраженностью сращения костей черепа между собой и рядом других аномалий. Голова из-за краниосиностоза приобретает характерную форму «трилистника» или «листка клевера», наблюдается значительная гипоплазия средней трети лица. По причине грубого нарушения формирования черепа данный тип синдрома Пфайффера характеризуется выраженной умственной отсталостью и рядом неврологических нарушений. Пороки развития конечностей сводятся к расширению первых пальцев, синдактилии, анкилозу локтевых суставов. Кроме того, синдром Пфайффера 2-го типа часто сопровождается различными пороками развития внутренних органов. Все вышеперечисленные нарушения могут стать причиной летального исхода в раннем возрасте.
Такое различие в клинической картине и прогнозе различных вариантов синдрома Пфайффера накладывает свой отпечаток на наследование этого заболевания. Так как 1-й тип, особенно в случае своевременного выявления и правильного лечения, характеризуется сохранением нормального уровня интеллекта, фертильности и выживаемости больных, возможна аутосомно-доминантная передача патологии потомству. Более тяжелые 2-й и 3-й типы чаще всего приводят к ранней смерти или глубокой инвалидизации больных, эти варианты синдрома Пфайффера возникают только в результате спонтанных мутаций.
Диагностика
Своевременная диагностика синдрома Пфайффера крайне важна, так как она позволяет вовремя составить схему паллиативного лечения, тем самым избежать осложнений и значительно улучшить качество жизни больного. Но это справедливо лишь в отношении 1-го типа заболевания, так как при других вариантах патологии комплекс пороков развития настолько тяжел, что практически не оставляет шансов на сохранение интеллекта, а в дальнейшем – жизни больных.
Для выявления синдрома Пфайффера используют ультразвуковые методики (в том числе и в качестве пренатальной диагностики), рентгенографию, молекулярно-генетические анализы. На профилактических УЗИ во время беременности при наличии этого заболевания у ребенка во 2-3 триместре можно выявить нарушение формирования черепа, синдактилию, пороки развития внутренних органов.
Рентгенологические методы редко используют при ранней диагностике синдрома Пфайффера у детей – в основном, производят исследование черепа, которое обнаруживает наличие краниосиностоза различной степени выраженности, гипоплазию верхнечелюстных костей, изменение формы глазниц. Также на рентгене можно определить изменение формы костей больших пальцев рук и ног.
Молекулярно-генетическая диагностика производится врачом-генетиком – в этом случае при подозрении на наличие синдрома Пфайффера 1-го типа производят поиск точечной мутации 252Pro-Arg методом прямого секвенирования экзона 7а гена FGFR1. В других случаях выполняют секвенирование 7-го и 9-го экзонов гена FGFR2 – именно там возникает большинство дефектов, приводящих к развитию синдрома Пфайффера. Вспомогательную роль в диагностике заболевания могут играть методы медицинской визуализации, направленные на определение аномалий развития внутренних органов.
Лечение синдрома Пфайффера
Лечение синдрома Пфайффера только симптоматическое и паллиативное – специалисты стараются устранить повышенное внутричерепное давление, обеспечить нормальное развитие нервной системы, снизить выраженность нарушений со стороны внутренних органов. Для этого широко применяют хирургические методики – разделение пальцев, пластику черепа и ряд других. Также назначают препараты, направленные на улучшение питания нервной ткани – ноотропные средства, витамины. При своевременно начатом лечении синдрома Пфайффера 1-го типа выживаемость больных значительно увеличивается. К сожалению, объем медицинской помощи зачастую недостаточен для полноценного лечения больных синдромом Пфайффера 2-го и 3-го типов.
Прогноз и профилактика
Прогноз синдрома Пфайффера определяется вариантом этого заболевания. Первый тип характеризуется относительно благоприятным прогнозом, который зависит от своевременности выявления патологии и правильности подобранного лечения. В благоприятных условиях больные сохраняют нормальный интеллект, доживают до преклонного возраста и способны иметь потомство – при этом риск передачи заболевания детям составляет минимум 50%.
Однако синдром Пфайффера 2-го и 3-го типов обладает крайне неблагоприятным прогнозом – совокупность неврологических и других нарушений часто становится причиной летального исхода в раннем детстве. Даже при выполнении паллиативных мероприятий у больных наблюдается выраженная олигофрения и глубокая инвалидизация. Профилактика синдрома Пфайффера, учитывая часто спонтанный механизм развития этого заболевания, возможна только в рамках пренатальной диагностики ультразвуковыми или генетическими методами.
От синдрома нет лекарства. Лечение является поддерживающим и часто включает в себя хирургическое вмешательство в первые годы жизни для исправления деформаций черепа и респираторной функции. Большинство людей с синдромом Пфайффера типа 1 имеют нормальный интеллект и продолжительность жизни, тогда как типы 2 и 3 обычно приводят к нарушениям развития нервной системы и ранней смерти.
Синдром Пфайффера встречается примерно у 1 из 100 000 новорожденных. Синдром назван в честь немецкого генетика Рудольфа Артура Пфайффера (1931–2012), который описал его в 1964 году.
СОДЕРЖАНИЕ
Признаки и симптомы
Многие характерные черты лица являются результатом преждевременного сращения костей черепа ( краниосиностоз ). Голова не может нормально расти, что приводит к высокому выступающему лбу ( туррибрахицефалия ) и глазам, которые кажутся выпуклыми ( проптоз ) и широко посаженными ( гипертелоризм ). Кроме того, имеется недоразвитая верхняя челюсть ( гипоплазия верхней челюсти ). Более 50 процентов детей с синдромом Пфайффера страдают потерей слуха; проблемы с зубами также распространены.
У людей с синдромом Пфайффера большие пальцы рук и пальцы ног широкие и отклоняются от других пальцев ( варусный поллекс и варусный палец стопы ). Также распространены необычно короткие пальцы рук и ног ( брахидактилия ), и между пальцами может быть перепонка или слияние ( синдактилия ).
Причина
Диагностика
Классификация
Наиболее широко принятая клиническая классификация синдрома Пфайффера была опубликована М. Майклом Коэном в 1993 году. Коэн разделил синдром на три, возможно, перекрывающихся типа, каждый из которых характеризуется широкими большими пальцами рук, широкими большими пальцами ног, брахидактилией и, возможно, синдактилией :
Управление
Ключевой проблемой является раннее сращение черепа, которое можно исправить с помощью ряда хирургических процедур, часто в течение первых трех месяцев после рождения. В дальнейшем необходимы операции по исправлению деформаций дыхательных путей и лица.
Итоги
Дети с синдромом Пфайффера типа 2 и 3 «имеют более высокий риск нарушений развития нервной системы и более низкую продолжительность жизни», чем дети с синдромом Пфайффера типа 1, но при лечении возможны благоприятные исходы. В тяжелых случаях респираторные и неврологические осложнения часто приводят к преждевременной смерти.
История
Синдром назван в честь немецкого генетика Рудольфа Артура Пфайффера (1931–2012). В 1964 году Пфайффер описал восемь человек в трех поколениях семьи, у которых были аномалии головы, рук и ног ( акроцефалосиндактилия ), унаследованные по аутосомно-доминантному типу.








ВРОЖДЕННОЕ ЗАБОЛЕВАНИЕ – СИНДРОМ ПФАЙФФЕРА
Синдром описан Pfeiffer в 1964 г., обнаружившим в трех родословных восемь случаев заболевания, основными клиническими проявлениями которого были краниосиностоз, широкие большие пальцы кистей и стоп, частичная синдактилия мягких тканей кистей рук. Для данного синдрома характерен аутосмонодоминантный тип наследования с полной пенетрант-ностью и вариабельной экспрессивностью. С Синдромом Пфайффера рождается один ребёнок на 100 000 новорождённых.
Cohen (1993) предложил три клинических подтипа синдрома, которые не имеют самостоятельного статуса, но имеют важное диагностическое и прогностическое значение: 1-й тип имеет аутосомно-доминантный тип наследования, мутации носят спорадический характер; 2-й и 3-й типы носят только спорадический характер.
Частота встречаемости
Анамнез. Синдром диагностируется в раннем возрасте за счет обнаружения деформации костей черепа и характерных аномалий конечностей.
Симптомы и проявления
Одной из главных особенностей синдрома Пфайффера является преждевременное слияние некоторых костей черепа (краниосиностоз). Это раннее слияние приводит к нарушениям в росте черепа и, как следствие, к аномальной форме головы и лица. При синдроме Пфайффера также часто отмечаются аномалии костей рук и ног.
Многие из характерных лицевых особенностей синдрома Пфайффера являются результатом преждевременного слияния костей черепа. У большинства детей отмечаются широко расположенные и выпуклые глаза, высокий лоб, слаборазвитая верхняя челюсть и нос в форме клюва. У более половины всех детей также отмечаются потеря слуха, стоматологические проблемы, аномалии костей конечностей (большие пальцы часто широкие, пальцы ног необычно короткие, в некоторых случаях отмечаются синдактилии)
Синдром Пфайффера подразделяется на три типа.
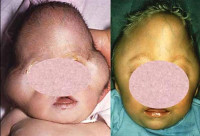
Лицо ребёнка с синдромом Пфайффера I типа:
грубый проптоз глазных яблок
выраженная брахицефалия с недоокостенением свода черепа
Кисти рук ребёнка с синдромом Пфайффера I типа:
лёгкая синдактилия III и IV пальцев правой кисти
Рентгенограмма стопы ребёнка с синдромом Пфайффера I типа:
укороченная и частично удвоенная первая плюсневая кость
отсутствие средних фаланг II-V пальцев
«2 и 3 типы» более серьёзные и связаны с проблемами нервной системы. Преждевременное «срастание» костей черепа может ограничить рост мозга, что может привести к задержке в развитии и другим неврологическим проблемам. Череп в виде листка клевера, экзорбитизм, широкие большие пальцы, анкилоз локтевого сустава, сниженный интеллект и серьезные нарушения со стороны ЦНС. Большинство больных с синдромом Пфайффера (Pfeiffer) 2-го типа умирают вскоре после рождения от дыхательной недостаточности, аномалий головного мозга, недоношенности или послеоперационных осложнений.
«2 тип» отличается от «3 типа» тем, что может привести к формированию головы в виде клеверного листа, причиной этого является обширное «срастание» костей черепа.
Лица детей с синдромом Пфайффера II типа:
череп в виде «трилистника»
Лицо ребёнка с синдромом Пфайффера III типа:
деформация черепа по типу «трилистника» отсутствует
грубый проптоз глазных яблок за счёт плоских глазниц
Стопа ребёнка с синдромом Пфайффера III типа:
Описан немецким медицинским генетиком Рудольфом Артуром Пфайффером в 1964г. (Pfeiffer R.A. Dominant erbliche Akrocephalosyndaktylie // Z.Kinderheilk., 1964. – Bd.90. – S.301-320
Клинические проявления
Черепно-лицевая область. Акроцефалия — основной и постоянный признак синдрома, но краниостеноз при этом отмечается не всегда. Возможен синостоз коронарного, сагиттального и ламбдовидного швов. В отдельных случаях наблюдается асимметрия черепа. Черепно-лицевые дисморфии сходны с таковыми при синдроме Аперта: уплощенная спинка носа, гипоплазия верхнечелюстных костей, гипертелоризм, антимонголоидный разрез глазных щелей, высокое арковидное небо, сверхкомплектные зубы. Вместе с тем каждый из названных признаков встречается реже, чем при синдроме Аперта, и выраженность их меньше. Расщелины неба встречаются в 14% случаев. Экзофтальм и косоглазие выявляются примерно стой же частотой.
В части случаев при синдроме Пфайффера отмечают необычную патологию черепа — в форме трилистника или листа клевера. В основе этого дефекта лежит преждевременный синостоз коронарного и ламбдовидного швов и развивающаяся внутриутробно гидроцефалия, в результате чего лобно-теменной отдел мозга как бы отделяется от боковых височных долей. В случае сочетания синдрома Пфайффера с аномалией черепа в форме трилистника, у больных обнаруживаются пороки развития мозга (микрополигирия, аплазия мозолистого тела, недоразвитие островков Гейла), патология глаз (колобомы радужки, афакия), сердца (аплазия створок аортального клапана), а также атрезия хоан и пилоростеноз.
Soekarman описал классический синдром Пфайффера у матери и сына. Сын имел форму черепа в виде трилистника. Развитие ребенка после хирургического лечения было нормальным, что доказывает: дети с формой черепа в виде трилистника могут иметь благоприятный прогноз.Конечности и скелет. Широкие пальцы на нижних конечностях являются классическим клиническим признаком синдрома Пфаифера. Изменения нижних конечностей при этом синдроме могут обладать значительной вариабельностью. Anderson в 1999 г. клинически и рентгенологически обследовал 22 пациента с синдромом Пфайффера. Только у 4-х из них рентгенологическая картина была нормальной.
Более выраженные изменения суставов были отмечены при синдромах Пфайффера и Аперта. При синдроме Сетре-Хотцена коленные суставы были нормальными. При синдроме Крузона наблюдались незначительные изменения. Наиболее тяжелыми аномалии суставов были у детей с пансиностозами, что в ряде случаев требовало хирургического вмешательства. У остальных — рентгенологические изменения отмечались не только в фалангах пальцев, но и в костях предплюсны и плюсны.
Снижение слуха было обнаружено у 8 из 9 пациентов. Степень снижения слуха была различна. 7 пациентов имели кондуктивную тугоухость и 1 — потерю слуха смешанного генеза. Сенсоневральное снижение слуха не было обнаружено ни у одного пациента. Четверо больных имели в анамнезе воспаление среднего уха. Первичные результаты компьютерной томографии (КТ) показали стеноз и/или атрезию наружного слухового канала, гипоплазию средней впадины уха. Слуховые косточки были гипоплазированы в нескольких случаях. Только в одном случае анатомия внутреннего уха была нормальна.
Этиология
Shotelersuk в 2002 г. описал 15-летнего тайского мальчика с неуказанным синдромом с краниосиностозом, характеризующегося мультишовным краниосиностозом, постоянным передним родничком, склерозом роговицы, стенозом хоан, атрезией слухового хода, широкими большими пальцами ног, выраженным сколиозом, acanthosis nigricans, гидроцефалией и умственной отсталостью. Рентген показал костный анкилоз тел позвонков в Т9-Т12, также как анкилоз костей кисти и фаланг пальцев рук и ног. Пациент оказался носителем мутации G870T, приводящей к замене W290C во внеклеточной области FGFR2. Как оказалось позже, у пациента был синдром Пфайффера.
Все известные случаи рождения пациентов с синдромом II типа были спорадическими, получившимися в результате новых мутаций в гене FGFR2 или в каком-то другом, не идентифицированном гене. Plomp в 1998 г. описал 5 пациентов (3 мальчика и 2 девочки) с синдромом II типа. Большинство пациентов с этой формой умерло вскоре после рождения. Причины смерти включали дыхательную недостаточность, патологию головного мозга, ранние и поздние осложнения. У двух пациентов обнаружили мутацию cys342arg.
Rossi в 2003 г. описал 4 семьи с общей мутацией гена FGFR1 P252R, однако не у всех носителей мутации череп был изменен. На ногах имелся широкий и сглаженный hallux, который обычно в середине отклоняется; кроме того, определялась синдактилия вторых и третьих пальцев ног. Авторы предположили, что эта характерная деформация ног, даже в случае отсутствия краниосиностоза, может быть основанием для поиска мутации P252R в FGFR1. При изучении спорадических случаев синдромов Крузона и Пфайффера, Glaser и соавт. в 2000 г. исследовали 4 внутригенных полиморфизма при скрининге 41 семьи. В 22 случаях (по 11 для каждого синдрома) полиморфизмы были информативны. Авторы нашли 11 различных мутаций в этой выборке. С помощью молекулярных методов было показано, что все мутации были отцовского происхождения. Больший возраст был отмечен у отцов пациентов с синдромом Крузона и Пфайффера по сравнению с отцами детей контрольной группы (34.50+/-7.65 лет против 30.45+/-1.28 год; р А. Разный фенотипический эффект данных мутаций скорее всего обусловлен тем, что вторая мутация не ведет к полному нарушению нормального сплайсинга премРНК FGFR 2.
В качестве примера, иллюстрирующего клинические проявления синдрома Пфайффера, рассмотрим выписку из истории болезни больного ребенка К. в возрасте 2 лет 8мес., находившегося на лечении в психоневрологическом отделении для детей раннего возраста № 4 городской детской больницы (ГДБ) г. Белгорода с 12.05 2015 г. по 18.05.2015 г. Больной ребенок 2012 года рождения поступил в отделение с жалобами на нарушение сна, беспокойство, задержку речи, повышенную потливость.
Из анамнеза жизни известно, что ребенок от 1 беременности, 1 оперативных родов путем кесарева сечения на 36 неделе беременности. Беременность у матери протекала на фоне хронической фетоплацентарной недостаточности (ХФПН), хламидиоза, анемии, токсикоза в виде тошноты в первой ее половине и нефропатии. Вес при рождении ребенка был 2240 г, рост – 45 см. Оценка по шкале Апгар – 7-8 баллов. К груди был приложен на 3 сутки, грудь не взял в виду отсутствия сосательного рефлекса. Кормление осуществлялось через зонд. Из роддома на 8 сутки после рождения был переведен в отделение патологии новорожденных (ОПН) областной детской клинической больницы (ОДКБ) г. Белгорода на дальнейшее обследование и лечение с диагнозом: перинатальное поражение центральной нервной системы смешанного генеза. Синдром Пфайффера? В ОПН был установлен и генетически подтвержден диагноз: синдром Пфайффера. На 9 день жизни ребенок был прооперирован в отделении детской хирургии ОДКБ по поводу синдрома Ледда. После выписки из отделения ребенок наблюдался в детской поликлинике у специалистов по месту жительства с диагнозом: перинатальное поражение центральной нервной системы смешанного генеза, синдром Пфайффера. Гипохромная анемия 1 степени. Ангиопатия сетчатки, субатрофия дисков зрительных нервов обоих глаз (ДЗН ОИ). Малые аномалии развития сердца (МАРС): открытое овальное окно (ООО), НК 0 ст. Состояние после операции по поводу синдрома Ледда. Тимомегалия 1 ст. Гипокальциемия. На грудном вскармливании находился до 1 года. Сроки прикормов и пищевых корригирующих добавок назначались согласно возраста по рекомендациям участкового педиатра. Аллергоанамнез не отягощен. В наследственном анамнезе у матери ребенка отмечается хронический пиелонефрит. Из перенесенных заболеваний до 1 года: ОРЗ, острый бронхит. В возрасте 1 года в феврале 2013 г. ребенок был впервые прооперирован по поводу краниостеноза в нейрохирургическом отделении Морозовской детской городской клинической больницы г. Москвы. Проведена реконструктивная краниопластика, установка металлоконструкций. Состояние после операции было удовлетворительным. Через полгода после краниопластики в плановом порядке проведено повторное оперативное вмешательство с целью удаления металлоконструкций в нейрохирургическом отделении Морозовской детской городской клинической больницы г. Москвы. 03.03.2014 г. проведено оперативное лечение: установка наружного люмбального дренажа. Реконструктивная краниопластика лобной кости. Пластика твердой мозговой оболочки (ТМО) лобной области. Операция прошла успешно. Состояние после операции удовлетворительное. По результатам компьютерной томографии головного мозга от 06.03.2014 г. отмечалось уменьшение размеров желудочков мозга по сравнению с исследованием от 11.02.2014 г. В правой лобной доле, в области порэнцефалической кисты, сообщающейся с передним рогом правого бокового желудочка, выявлялись гематома до 1.5 см в диаметре, признаки пневмоцефалии. Срединные структуры – не смещены. Срединно и справа в лобнотеменной и лобновисочной области прослеживались титановые сетки, скрепляющие костные лоскуты. После операции на фоне ОРЗ у ребенка появился кашель. В общем анализе крови отмечался лейкоцитоз до 17.7×109/л, ускорение СОЭ до 17 мм/час. Со стороны других лабораторных исследований крови и мочи патология не была обнаружена. Педиатр диагностировал острый бронхит, очаговое поражение легких? Рентгенологическое исследование органов грудной клетки показало неоднородное затемнение в нижне- медиальных отделах слева на уровне отрезков 4-5 ребер. 17.03.2014 г. послеоперационные швы были сняты. Воспалительных изменений в области послеоперационного рубца не было. Однако, учитывая физикальные изменения в легких, усиление кашля, данные лабораторно-инструментальных исследований, ребенок был переведен на дальнейшее обследование и лечение в педиатрическое отделение, где он лечился с диагнозом: сепсис, двусторонняя полисегментарная деструктивная пневмония, двусторонний гидроторакс. Задержка речевого развития, миотонический синдром. Гипохромная анемия 1 ст., тимомегалия 1ст. Ангиопатия сетчатки. Из отделения был выписан по месту жительства в удовлетворительном состоянии с соответствующими рекомендациями под наблюдение участкового педиатра. В течение последующего времени ребенок регулярно наблюдался у невролога, генетика, ортопеда, логопеда и педиатра по месту жительства. В двигательной сфере отмечалась положительная динамика: ребенок ходит самостоятельно, объем активных и пассивных движений полный. Однако, учитывая появившиеся жалобы на затруднение движений в пальцах верхних конечностей, беспокойный сон, ребенок был госпитализирован в психоневрологическое отделение для детей раннего возраста № 4 ГДБ г. Белгорода для контрольного обследования и лечения, где находился на госпитализации с 11.08.2014 г. по 26.08.2014 г. с диагнозом: резидуально–органическое поражение ЦНС на фоне синдрома Пфайффера. Краниосиностоз, состояние после повторной реконструкции. Аффективно- респираторный синдром. Пирамидная недостаточность в ногах. Задержка психоречевого развития. Ринит. В отделении ребенок получил следующее лечение: внутрь – диакарб, аспаркам, фенибут, сонапакс; внутримышечно – актовегин, мексидол; физиолечение (массаж общеукрепляющий, лазеротерапия на речевые зоны), капли в нос. После выписки из отделения у ребенка наблюдалась незначительная положительная динамика со стороны психоречевого развития. В последующем выполнялись рекомендации невролога отделения по дальнейшему лечению и наблюдению под контролем педиатра и детского невропатолога по месту жительства. Однако за последнее время у ребенка отмечается нарастание проявлений аффективно-респираторного синдрома. Аффективно-респираторные пароксизмы чаще возникают в ответ на эмоциональное возбуждение, провоцируются плачем, негативными проявлениями на осмотр врача и на проведение медицинских манипуляций. С целью дообследования ребенка, определения динамики основного патологического процесса и исключения очаговых и диффузных изменений мозга ребенок был вновь госпитализирован в психоневрологическое отделение для детей раннего возраста № 4 ГДБ 12.05.2015 г. с жалобами на повышенное беспокойство, повышенную потливость, нарушение сна. При поступлении в отделение общее состояние ребенка средней степени тяжести, обусловленное неврологической симптоматикой. Кожные покровы бледные, чистые. При осмотре определяются лицевые стигмы дизэмбриогенеза (СДЭ): акроцефалия, плоское лицо, антимонголоидный разрез глаз, выраженный экзоорбитизм, гипертелоризм глаз, сходящееся косоглазие, проптоз глазных яблок, диспластичные низко посаженные уши, гипопластический короткий плоский нос с поднятым широким кончиком и ноздрями, глубокая переносица, эпикант, гипоплазия верхней челюсти с выступающим альвеолярным отростком, прогнатия, высокое «готическое» небо, неправильный рост зубов, короткая шея с низким ростом волос. Также определяются СДЭ со стороны верхних и нижних конечностей: брахидактилия, пальцы верхних конечностей короткие и толстые, широкие большие пальцы стоп, вальгусная деформация стоп. Грудная клетка при осмотре деформирована. Частота дыхания (ЧД) – 24 в минуту. Перкуторно – звук ясный легочный с обеих сторон. При аускультации – дыхание в легких пуэрильное, хрипов нет. Область сердца при осмотре не изменена. Границы относительной тупости сердца при перкуссии в норме. При аускультации тоны сердца приглушены, ритмичные. Частота сердечных сокращений (ЧСС) – 110 ударов в минуту. При осмотре на передней брюшной стенке определяется послеоперационный рубец. Живот мягкий, безболезненный при пальпации. Печень выступает из-под края реберной дуги на 1.0 см. Селезенка не выступает из-под края левой реберной дуги. Почки не пальпируются. Стул и мочеиспускание – без патологических особенностей.
Неврологический статус: ребенок активен, сознание ясное. На осмотр реагирует негативно. Общемозговых, менингеальных симптомов нет. Череп «башенной» формы. Глазные щели симметричны, движения глазных яблок вверх, вниз в полном объеме. Зрачки округлой формы, S=D. Фотореакция сохранена. Точки выхода тройничного нерва безболезненны. Нижняя челюсть по средней линии. Корнеальный и конъюнктивальный рефлексы сохранены. Лицо симметричное.
Слух ориентировочно не нарушен. Нистагма нет. При глотании не поперхивается. Глоточный и небный рефлексы оживлены. Гиперсаливация. Голова по средней линии. Язык в полости рта по средней линии. Фасцикуляций и фибрилляций нет. В двигательной сфере: ходит самостоятельно. Отмечается атаксия. Объем активных и пассивных движений полный. Атонии мышц нет. Контрактуры отсутствуют. Дистония. Отмечается нарушение мелкой моторики. Сухожильно-периостальные рефлексы с конечностей оживлены. Со стороны психоэмоциональной сферы: ребенок активный, эмоционально беден. В контакт вступает ситуативно, избирательно. Задержка психоречевого развития. Уровень развития ниже возрастной нормы. Отмечается моторная алалия с элементами эхолалии.
Риск рецидива.
Различный, зависит от этиологии. В тех случаях, когда заболевания наследуются аутосомно-доминантно, то риск составляет 50%. Если же синдром возникает вследствие новых мутаций, то риск рецидива очень низок.
Патогенез
Нарушения со стороны внутренних органов представлены патологией костей таза, соха valga, пилоростенозом, пупочной грыжей. К редким аномалиям, встречающимся при синдроме Пфайффера (Pfeiffer), можно отнести стеноз трахеи, вызванный заменой ее хрящевых колец на хрящевую пластину.
Синдром Пфайффера (Pfeiffer) вызывается мутацией генов FGFR1 и FGFR2 (I тип этого синдрома связан с мутациями в генах FGFR1 и FGFR2,типы II и III связаны с мутациями в гене FGFR2) и значительно связан с мутациями Фактора роста фибробластов 1 и 2. Эти рецепторы очень важны для правильного развития костей. Неожиданным было обнаружение идентичных мутаций в различных семейных случаях «истинных» синдромов Пфайффера (Pfeiffer) и Крузона (Crouzon) с различным фенотипом.
Мутации в данном гене приводят также к развитию: синдрома Аперта, синдрому морщинистой кожи Беаре-Стевенсона, синдрома дисплазии Bent bone, краниофациально-скелетно-дерматологической дисплазии, неспецифическому краниосиностозу, синдрому Джексона-Вейсса, синдрому LADD, синдрому Антли-Бикслера, синдрому Крузона, синдрому Сетре-Чотзена, скафоцефалии и аномалии Аксенфельда-Ригера, синдрому скафоцефалии, западения верхней челюсти и умственной отсталости, соматическая мутация приводит к раку желудка.
Литература
1. Козлова С. И., Демикова Н. С. Наследственные синдромы и медико- генетическое консультирование. – М.: КМК, 2007 – 448 с.
2. Кеннет Л. Джонс «Наследственные синдромы по Девиду Смиту» Атлас-справочник. Москва, Практика, 2011.
3. Naveh, Y., Friedman, A. Pfeiffer syndrome: report of a family and review of the literature. J. Med. Genet. 13: 277-280, 1976.
5. Koltunov D.E. 2010. Sindrom Pfayffera: klinicheskie proyavleniya i etiologiya. Voprosy diagnostiki v pediatrii [Pfeiffer syndrome: clinical manifestations and etiology]. (3), 3:42-46 (in Russian).
6. Koltunov D.E., Bel’chenko V.A. 2012. Kharakteristika skeletnykh deformatsiy u patsientov s sindromami Aperta, Kruzona, Pfayffera. Voprosy prakticheskoy pediatrii [The characteristic of skeletal deformations at patients with Apert, Kruzona, Pfeiffer syndromes]. (6), 6:57-62 (in Russian).
7. Koltunov D.E., Bel’chenko V.A. 2013. Diagnostika sindromal’nykh form kraniosinostozov. Voprosy prakticheskoy pediatrii. [Diagnosis of craniostenosis sуndrom forms]. (3), 3:52-55 (in Russian).
8. Rossi M., Jones R. L., Norbury G. et al. 2003. The appearance of the feet in Pfeiffer syndrome caused by FGFR1 P252R mutation. Clin. Dysmorph. 12:269-274.
9. Vogels A, Fryns JP. Pfeiffer syndrome. 2006. Orphanet Journal of Rare Diseases, 1:19.