Синдром протея что протея
Врожденное отсутствие отдельных мышц встречается достаточно часто и обычно носит асимметричный характер. Нередко наблюдается аплазия длинной ладонной мышцы на передней поверхности предплечья. Эта мышца отсутствует у 30 % здоровых людей, ее функция полностью компенсируется функцией других сгибателей запястья.
Одностороннее отсутствие грудинно-ключично-сосцевидной мышцы — одна из причин врожденной кривошеи. Отсутствие большой грудной мышцы служит компонентом синдрома Poland. При изменении иннервации мышц, например мышц нижних конечностей при тяжелых формах миеломенингоцеле, возможно нарушение развития мышц. При сакральной аге-незии аномалия сомитов приводит к дефекту формирования позвонков, что может служить причиной патологии мышц, развивающихся из той же аномальной мезодермальной пластинки; нарушение индукции вызывает сегментарную амиоплазию.
При отсутствии формирования длинных костей не происходит дифференцировки скелетных мышц конечностей из эмбриональных миомеров. Отсутствие одной длинной кости, такой как лучевая кость, ассоциируется с вариабельной степенью аплазии или гипоплазии соответствующих мышц, таких как лучевой сгибатель кисти. Конечную стадию неврогенной атрофии мышц иногда называют амиоплазией, хотя это название терминологически неточно.

Генерализованная амиоплазия обычно приводит к смерти плода; родившиеся живыми новорожденные редко выживают. Генетические исследования, проводимые на мышах, позволяют предположить, что в основе заболевания лежит мутация в одном из миогенных генов, однако в настоящее время это предположение не подтверждено в исследованиях на людях.
Мышечная дисгенезия (синдром Протея)
Синдром Протея — это нарушение клеточного роста с вовлечением эктодермальной и мезодермальной ткани. Причина заболевания неизвестна, однако синдром Протея не подчиняется менделевскому типу наследования. Заболевание проявляется в виде асимметричного избыточного роста конечностей, бородавчатых изменений на коже, ангиом различных типов, утолщения костей, гемимегалэнце-фалии и избыточного роста мышц при отсутствии мышечной слабости. При гистологическом исследовании мышц характерна мышечная дисгенезия.
Аномальные зоны прилегают к зонам нормально сформированных мышц и не соответствуют анатомическим границам. Заболевание может быть вызвано патологией секреторных ростовых факторов. Историческим примером заболевания служит «человек-слон», известный своим гротескным обликом, живший в Лондоне в конце XIX в. Этот человек вызвал настоящую сенсацию в обществе. Долгое время считалось, что он страдает нейрофиброматозом, однако в настоящее время этот случай расценивается как синдром Протея.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
— Вернуться в оглавление раздела «неврология»
Синдром Протея
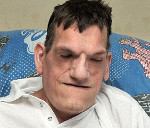
Синдром Протея – это редкая врожденная патология, проявляющаяся аномальным ростом отдельных частей тела. Обычно выявляется увеличение участка головы, руки или ноги. Частичная гипертрофия сочетается с повышенной вероятностью развития тромбозов и тромбоэмболий, образования опухолей различной структуры и локализации. Диагноз выставляется на основании результатов осмотра, данных рентгенографии, КТ, МРТ, ангиографии, допплерографии. Радикальное лечение не разработано. Применяются ортопедические приспособления, по показаниям осуществляются операции для улучшения функций конечности, облегчения дыхания и приема пищи, выполняется хирургическое удаление неоплазий.
МКБ-10

Общие сведения
Синдром Протея (парциальный гигантизм) – очень редкое врожденное заболевание. В литературе упоминается примерно 200 случаев данной патологии, при этом специалисты не исключают, что некоторые пациенты могли страдать другими болезнями со сходной симптоматикой. Первое описание синдрома было представлено в 1979 году, однако широкую известность он получил после публикации работы немецкого педиатра Видемана в 1983 году. Название также было предложено Видеманом по ассоциации с древнегреческим богом Протеем, способным к изменению внешнего облика. Заболевание оказывает выраженное негативное влияние на качество жизни больных, как из-за внешнего обезображивания, так и вследствие нарушений функций пораженных частей тела. Нередко становится причиной ранней смерти из-за высокой вероятности развития опухолей и склонности к тромбозам.
Причины
Причиной развития заболевания является соматический мозаицизм, предположительно обусловленный случайной мутацией доминантного гена. Некоторые специалисты подвергают сомнению случайный характер мутации, опираясь на информацию о выявлении нескольких случаев легких признаков синдрома у родителей больных. Существуют исследования, согласно которым патология обусловлена мутацией гена PTEN, расположенного в локусе 10q X хромосомы. Вместе с тем, исследователи указывают, что мутации данного гена обнаруживаются всего у 20% пациентов с синдромом Протея и определяются при некоторых других заболеваниях, характеризующихся повышенной вероятностью образования злокачественных неоплазий. Таким образом, генетическая основа болезни пока окончательно не установлена.
Патогенез
В результате мутации в отдельных частях тела образуется три вида клеток – нормальные, гипер- и атрофические. На измененном участке в процесс вовлекаются кожные покровы, костная, мышечная и жировая ткань, лимфатические и кровеносные сосуды, что вызывает деформацию, увеличение длины и объема конечности или части головы, изменения сосудов. Поскольку дефектный ген является супрессором опухолей, наличие мутации резко повышает вероятность появления новообразований.
Симптомы
При рождении признаки заболевания отсутствуют. Патологические проявления возникают в раннем детском возрасте. Клиническая картина полиморфная. Характерными симптомами являются аномальное увеличение одной или нескольких конечностей, макроцефалия, сосудистые аномалии (мальформации, варикозное расширение вен). У некоторых больных выявляются офтальмологические нарушения: экзофтальм, косоглазие, миопия. При патологическом росте нижней челюсти формируется прогения. Возможно разрастание кожи в области подошв. В числе возможных вариантов патологии в литературных источниках упоминается изолированное увеличение селезенки или пальцев рук, атрофия зрительного нерва и пигментная дегенерация сетчатки в сочетании с множественными менингиомами.
Увеличение нижних конечностей влечет за собой нарушение опоры и движений. Пациенты вынуждены пользоваться ортопедической обувью и специальными приспособлениями (тростью, костылями, инвалидной коляской). При увеличении верхних конечностей наблюдаются трудности в процессе самообслуживания. Из-за деформации частей головы возникают грубые косметические дефекты, могут отмечаться затруднения при дыхании и приеме пищи. Существует высокая вероятность развития новообразований различной локализации и гистологической структуры: гамартом, липом, лимфангиом, гемангиом, аденом слюнных желез, менингиом и других.
Осложнения
Типичным признаком болезни Протея является возникновение злокачественных опухолей, существенно сокращающих продолжительность жизни больных. Другими опасными для жизни последствиями заболевания считаются осложнения сосудистых патологий. Отмечается повышенный риск тромбоза глубоких вен. Описаны случаи развития тромбоэмболии легочной артерии. Из-за увеличения и разницы в длине нижних конечностей возрастает нагрузка на суставы и позвоночник, формируется искривление позвоночного столба. Обнаруживаются вторичные артрозы, остеохондроз. Более чем у половины пациентов выявляется умственная отсталость, обусловленная сдавлением нервных тканей увеличенными костями черепа. Возможны судороги, неврологические расстройства.
Диагностика
Из-за незначительной распространенности, сходства синдрома Протея с некоторыми другими врожденными заболеваниями постановка диагноза может представлять значительные затруднения. В процессе диагностического поиска принимают участие специалисты в области онкологии, травматологии и ортопедии, сосудистой хирургии и пр. В качестве основных критериев рассматривают гемигипертрофию, сосудистые аномалии и склонность к образованию опухолей. План обследования включает:
При нарушениях со стороны органа зрения назначается офтальмологическое обследование, при умственной отсталости применяются тесты для определения уровня интеллекта, когнитивных способностей. При судорожном синдроме, расстройствах деятельности центральной и периферической нервной системы показана консультация невролога. Заболевание дифференцируют с врожденным липоматозом и синдромами Клиппеля-Треноне-Вебера, Маффучи, Баннайана-Зоннана, при которых также выявляются опухоли, частичная гипертрофия и патология сосудов.
Лечение синдрома Протея
Патогенетическая терапия не разработана, лечение только симптоматическое. По показаниям осуществляют ортопедическую коррекцию с использованием специальной обуви, ортезов, корсетов при вторичных изменениях позвоночника. Назначают медикаментозную терапию для профилактики тромбозов. Из-за вовлечения различных органов и систем оперативные лечебные мероприятия проводятся разными специалистами, в том числе – ортопедами, онкологами, нейрохирургами, вертебрологами, сосудистыми хирургами, хирургами-косметологами, челюстно-лицевыми хирургами и т. д. Можно выделить следующие группы операций при синдроме Протея:
Прогноз и профилактика
Синдром Протея является инвалидизирующим заболеванием, негативно влияет на продолжительность жизни пациентов. По данным исследований, многие больные погибают в детстве или молодом возрасте. Причинами летального исхода становятся злокачественные неоплазии, ТЭЛА, тромбозы крупных сосудов. Профилактические мероприятия не разработаны из-за врожденного характера патологии и недостаточной изученности этиологии болезни. Пациентам, страдающим данным заболеванием, необходимо регулярно проходить обследование для оценки состояния свертывающей системы крови, раннего выявления новообразований.
Синдром Протея, или парциальный гигантизм

Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.

Редкое заболевание – синдром Протея – это генетическая мультисистемная патология с выраженными клиническими проявлениями, а именно: с выборочным гигантизмом, поражением системы крово и лимфотока.
Первое упоминание о синдроме принадлежит Майклу Коэну – датировано оно 1979 годом. Через четыре года в Германии патология получила свое нынешнее название – синдром Протея, по имени древнегреческого божества Протея-многоликого.
Часто синдром Протея ошибочно диагностируют, как гамартоматозную болезнь в виде неврофиброматоза, которая передается аутосомно-доминантно.
Другие названия синдрома Протея: болезнь многоликости, заболевание человека-слона, синдром парциального гигантизма.
 [1], [2], [3], [4], [5], [6]
[1], [2], [3], [4], [5], [6]
Код по МКБ-10
Эпидемиология
Синдром Протея считается крайне редкой патологией, так как за все время было обнаружено лишь около двух сотен подобных случаев. Такая статистика позволяет утверждать, что синдром возникает менее чем в 1 случае на 1 млн. новорожденных детей.
[7], [8], [9], [10]
Причины синдрома Протея
Причина развития синдрома Протея состоит в определенной генной мутации. Все мы знаем, что человеческий организм имеет большое количество ДНК-цепочек, которые передаются нам от родителей. Но иногда во время эмбрионального развития происходит некий генетический сбой, что и приводит к появлению врожденных генетических заболеваний.
Синдром Протея обнаруживается при измененном гене АКТ: данный белок ответственен за скорость клеточного роста в организме. Если человек здоров, то его ген АКТ находится в неактивном состоянии. У больных синдромом Протея этот ген активен и ускоряет процесс роста клеток.
Тяжесть проявления патологии зависит от того, на каком именно внутриутробном этапе имела место мутация гена. Чем более ранним был этот этап, тем серьезнее будет протекать синдром Протея.
На данный момент ученые работают над такими вопросами:
[11], [12], [13], [14], [15], [16], [17], [18]
Факторы риска
Точные факторы, которые влияют на развитие синдрома Протея, неизвестны. Однако ученые выделили ряд факторов, которые теоретически могут способствовать появлению у ребенка такой патологии:
[19], [20], [21], [22], [23], [24]
Патогенез
Патогенез синдрома Протея до конца пока не изучен. Известно лишь, что к развитию патологии приводит мозаицизм соматических клеток – аномалия сочетания половых хромосом по доминантному гену, который до настоящего времени не определен.
Тем не менее, и эта теория некоторыми учеными ставится под сомнение, так как имелись одиночные случаи незначительных признаков заболевания у родителей пораженных индивидов.
Сочетанное существование гипер и гипоплазии при синдроме Протея наводит на мысль о возможной эмбриональной соматической рекомбинации, которая становится причиной появления не менее трех клеточных подвидов: нормальных, гипертрофических и атрофических клеточных структур.
[25], [26], [27], [28], [29]
Симптомы синдрома Протея
Обычно люди, больные синдромом Протея, в младенческом возрасте не отличаются от других детей: патологические изменения начинают проявляться с годами. Диагностировать синдром Протея поначалу очень сложно, так как первые признаки болезни у каждого больного могут отличаться. Единственным характерным признаком заболевания является разрастание тканей. При этом разрастаться может любая ткань человеческого организма: костная, мышечная, жировая ткань, а также сосуды системы кровообращения и лимфотока. Разрастание может поразить практически любой орган. Правда, больший процент разросшейся ткани фиксируется на конечностях и в области головы.
Синдром Протея напрямую влияет на снижение продолжительности жизни больного, и это объясняется частыми проблемами с сосудами. Среди подобных проблем обычно встречаются тромбоэмболии, тромбозы и т. п. Не менее распространены опухолевые процессы и поражения эндокринной системы.
Сам по себе синдром не оказывает влияния на снижение интеллектуального уровня больного, но патологическое разрастание нервной ткани может спровоцировать значительное отставание умственного развития.
Первые признаки синдрома могут проявляться у детей, начиная с 2-х или 4-х лет. Обычно это такие симптомы:
Осложнения и последствия
[30], [31], [32], [33], [34], [35], [36], [37]
Диагностика синдрома Протея
Диагностировать синдром Протея на начальной стадии практически невозможно, так как основными критериями диагностики являются характерные симптомы заболевания:
Анализы для подтверждения диагноза синдрома Протея не проводятся. Обязательно контролируют показатели свертываемости крови, так как для болезни характерны тромбозы и тромбоэмболии.
Инструментальная диагностика может включать в себя рентгенологическое исследование, магнитно-резонансную томографию, компьютерную томографию, ангиографию, энцефалографию и пр.
Иногда при синдроме Протея, особенно при наличии опухолевых процессов, врачи назначают гистологическое исследование с предварительной биопсией.
[38]
Дифференциальная диагностика
Дифференциальная диагностика синдрома Протея проводится со следующими заболеваниями:
[39], [40], [41], [42], [43], [44], [45], [46]
К кому обратиться?
Лечение синдрома Протея
Синдром Протея считается неизлечимым заболеванием. Однако ранняя диагностика болезни позволяет успешно побороть основные признаки патологии и избежать осложнений. Например, при искривлении позвоночного столба, при избыточном росте костной ткани, при несоответствии длины конечностей больному предлагается применять специальные ортопедические приспособления.
Если же расстройство наблюдается в системе кроветворения, либо обнаруживается рост опухолевых процессов, то больной синдромом Протея должен находиться под пожизненным врачебным наблюдением.
Медикаментозное лечение синдрома Протея состоит только лишь в назначении симптоматических препаратов. К ним относятся обезболивающие средства (Ибупрофен, Кетолонг), мочегонные препараты (Фуросемид, Лазикс), антикоагулянты (Гепарин, Фрагмин, Фондапаринукс, Тинзапарин), вазопрессоры (Допамин, Добутамин), тромболитики (Урокиназа, Стрептокиназа, Альтеплаза).
Лекарства, допускаемые к применению при синдроме Протея
Способ применения и дозы
При болях принимают по 600 мг 2-3 раза в сутки.
Прием может сопровождаться тошнотой, диспепсией, болями в желудке.
Ибупрофен не применяют при нарушении кроветворной функции.
При отеках принимают 20-80 мг в день, с возможным дальнейшим увеличением дозировки.
Возможно понижение давления, слабость, боли в голове, жажда, аллергия.
Прием препарата должен сочетаться с компенсацией электролитных нарушений.
Применяется в качестве антикоагулянта по индивидуальным схемам лечения.
При длительном лечении возможно развитие геморрагических осложнений.
Препарат применяют, постоянно контролируя степень свертываемости крови.
Препарат применяют по индивидуально подобранным схемам.
Длительное лечение может стать причиной появления аритмии, изменения артериального давления.
При лечении препаратом необходимо контролировать ЧСС, кровяное давление, диурез.
Препарат назначают внутривенно капельно, в средней дозировке 250 000 МЕ в 50 мл физраствора со скоростью 30 кап./мин.
Возможна гиперреакция на белок: боль в голове, тошнота, лихорадка.
Лечение проводят с контролем показателей свертываемости крови и уровня фибриногена.
Витамины
Рацион человека, страдающего синдромом Протея, должен включать в себя витаминизированные и сбалансированные по составу блюда. Кроме этого, периодически можно принимать дополнительные витамины – в основном, для укрепления сосудов, сердца, для улучшения состояния и структуры клеток и тканей.
Витаминные препараты обычно назначают индивидуально, так как у некоторых пациентов возможна непереносимость того или иного компонента.
Физиотерапевтическое лечение
Физиотерапия при синдроме Протея обычно направлена на поддержание функции сердечно-сосудистой системы, улучшение коронарного и периферического кровообращения. С улучшением циркуляции крови повышается уровень транспортировки кислорода, облегчается протекание процессов в центральной нервной системе и вегетативной нервной системе, нормализуются нейроэндокринные и иммунные реакции.
Для улучшения состояния пациента с синдромом Протея могут использоваться различные физиотерапевтические методы, в зависимости от доминирующего проявления заболевания.
Противопоказаниями к физиотерапии могут стать:
Выбор конкретной процедуры при синдроме Протея зависит от выраженности функционального расстройства сердечно-сосудистой системы, от состояния нервной и нейрогуморальной системы регуляции кровообращения, а также от наличия других неполадок в организме.
Народное лечение
Для улучшения качества крови при синдроме Протея рекомендуется регулярно употреблять напитки из ягод калины, облепихи, клюквы, черники.
Для профилактики тромбообразования при синдроме Протея полезными считаются чаи и настои на основе мать-и-мачехи, таволги, астрагала, окопника, листьев малины. Заваривают 1 ст. л. травы в ½ л кипящей воды, настаивают под крышкой до остывания. Такое лекарство пьют по половине стакана три раза в сутки.
Огромную пользу оказывает знаменитый имбирный чай: он разжижает кровь и улучшает кровообращение, препятствует скоплению токсинов. Чтобы приготовить лечебный имбирный чай, кусочек имбирного корня натирают на терке и заливают кипятком, настаивают 20 минут. После остывания в напиток добавляют немного меда и/или лимона. Для улучшения лимфотока в такой чай можно добавить щепотку корицы.
Если в пораженных тканях образовались отеки, то используют такой способ: к пораженным участкам прикладывают нарезанный на дольки томат: через 3-4 часа дольки заменяют свежими.
Пример лечения синдрома Протея при помощи мумиё:
Курс лечения можно провести повторно через 5 дней. Всего рекомендовано 4 курса.
Пораженные участки можно обрабатывать мазью, которая представляет собой 20% разведенное мумиё в смеси с вазелином.
[47], [48], [49], [50], [51], [52], [53], [54], [55], [56], [57]
Лечение травами
Гомеопатия
На сегодняшний день многие медицинские специалисты признали эффективность гомеопатических препаратов в лечении различных, в том числе и хронических, заболеваний. Существуют и такие гомеопатические средства, которые могут помочь, если не вылечить, то значительно ослабить проявления такой болезни, как синдром Протея.
К примеру, Лимфомиозот – это комплексный гомеопатический препарат немецкого производства, который выполняет сразу несколько полезных функций в организме:
Лимфомиозот можно приобрести в аптеках в виде капель, таблеток или инъекционного раствора. Препарат рекомендован к применению по предписанию доктора трижды в сутки (таблетки или капли), либо 1-3 раза в неделю (внутримышечные или подкожные инъекции). Курс лечения Лимфомиозотом может продолжаться долго, при необходимости до нескольких месяцев.
Для того, чтобы усилить эффективность препарата, его можно сочетать с другими гомеопатическими средствами. Чаще всего используют такие препараты: кониум, туя, кальциум флюорикум. Реже и по показаниям – солянум туберозум, сукцинум, апис и тропеолум.
Кроме этого, в течение последнего десятилетия активно используют потенцированные кейлоновые средства (препараты-регуляторы митотического клеточного деления), средства эпидермального фактора роста (EGF), средства фактора роста фибробластов (FGF).
Перечисленные препараты практически лишены неприятных побочных эффектов, однако могут существенно улучшить состояние больных синдромом Протея.
Оперативное лечение
Некоторые виды тканевых разрастаний при синдроме Протея требуют оперативной коррекции. Например, при челюстных деформациях приводятся:
При наличии кожных и подкожных разрастаний, поверхностных гемангиом, может применяться их лазерное удаление или криодеструкция. Кистозные образования и опухоли (в том числе и внутренние) удаляют оперативным путем.
Некоторые операции при синдроме Протея проводятся по эстетическим соображениям – к примеру, если разрастание ткани обнаруживается в области лица или головы.
Синдром Протея у ребенка 14 лет 11 месяцев
Полный текст:
Аннотация
Ключевые слова
Об авторах
канд. мед. наук, ассистент кафедры педиатрии факультета повышения квалификации и профессиональной переподготовки специалистов,
410012, г. Саратов, ул. Большая Казачья, 112
д-р мед. наук, профессор, заведующий кафедрой педиатрии факультета повышения квалификации и профессиональной переподготовки специалистов,
410012, г. Саратов, ул. Большая Казачья, 112
д-р мед. наук, профессор кафедры педиатрии факультета повышения квалификации и профессиональной переподготовки специалистов,
410012, г. Саратов, ул. Большая Казачья, 112
канд. мед. наук, доцент кафедры педиатрии факультета повышения квалификации и профессиональной переподготовки специалистов,
410012, г. Саратов, ул. Большая Казачья, 112
канд. мед. наук, ассистент кафедры педиатрии факультета повышения квалификации и профессиональной переподготовки специалистов,
410012, г. Саратов, ул. Большая Казачья, 112
врач высшей категории, заведующая педиатрическим отделением,
413100, Саратовская область, г. Энгельс, пл. Свободы, 23
Список литературы
1. Кузнецова МА, Зайцева ГВ, Зрячкин НИ, Макарова ОА, Хмилевская СА. Синдром Патау. Вопросы практической педиатрии. 2015;10(6):90–3.
2. Biesecker L. The challenges of Proteus syndrome: diagnosis and management. Eur J Hum Genet. 2006;14(11):1151–7. doi: 10.1038/sj.ejhg.5201638.
3. Biesecker LG. The multifaceted challenges of Proteus syndrome. JAMA. 2001;285(17):2240–3. doi: 10.1001/jama.285.17.2240.
4. Cohen MM Jr. Proteus syndrome review: molecular, clinical, and pathologic features. Clin Genet. 2014;85(2):111–9. doi: 10.1111/cge.12266.
5. Cohen MM Jr, Hayden PW. A newly recognized hamartomatous syndrome. Birth Defects Orig Artic Ser. 1979;15(5B):291–6.
6. Wiedemann HR, Burgio GR, Aldenhoff P, Kunze J, Kaufmann HJ, Schirg E. The proteus syndrome. Partial gigantism of the hands and/or feet, nevi, hemihypertrophy, subcutaneous tumors, macrocephaly or other skull anomalies and possible accelerated growth and visceral affections. Eur J Pediatr. 1983;140(1): 5–12.
7. Benyan AK, Fatehala ZF, AL-Hassani FAA. Proteus syndrome: a case report in Basra Iraq. Kufa Medical Journal. 2011;14(2):28–33.
8. Семячкина АН, Новиков ПВ, Воинова ВЮ, Курбатов МБ, Синельщикова ТА, Кузьмина НС, Засухина ГД. Синдром Протея у детей: диагностика, лечение и профилактика. Российский вестник перинатологии и педиатрии. 2007;52(1):45–9.
9. Ильина ЕГ, Ершова-Павлова АА, Бойша АС, Сапожникова ТВ, Фильчакова АМ, Селиванова ЛН, Шумская ЕА, Белозорова ТИ. Тяжелая форма синдрома Протея у новорожденного. Медицинская генетика. 2009;8(7): 39–41.
10. Lindhurst MJ, Sapp JC, Teer JK, Johnston JJ, Finn EM, Peters K, Turner J, Cannons JL, Bick D, Blakemore L, Blumhorst C, Brockmann K, Calder P, Cherman N, Deardorff MA, Everman DB, Golas G, Greenstein RM, Kato BM, Keppler-Noreuil KM, Kuznetsov SA, Miyamoto RT, Newman K, Ng D, O’Brien K, Rothenberg S, Schwartzentruber DJ, Singhal V, Tirabosco R, Upton J, Wientroub S, Zackai EH, Hoag K, Whitewood-Neal T, Robey PG, Schwartzberg PL, Darling TN, Tosi LL, Mullikin JC, Biesecker LG. A mosaic activating mutation in AKT1 associated with the Proteus syndrome. N Engl J Med. 2011;365(7):611–9. doi: 10.1056/NEJMoa1104017.
11. Zhou X, Hampel H, Thiele H, Gorlin RJ, Hennekam RC, Parisi M, Winter RM, Eng C. Association of germline mutation in the PTEN tumour suppressor gene and Proteus and Proteus-like syndromes. Lancet. 2001;358(9277): 210–1. doi: http://dx.doi.org/10.1016/S0140-6736(01)05412-5.
12. Nguyen D, Turner JT, Olsen C, Biesecker LG, Darling TN. Cutaneous manifestations of proteus syndrome: correlations with general clinical severity. Arch Dermatol. 2004;140(8): 947–53. doi: 10.1001/archderm.140.8.947.