Моуат-Вильсон синдром
OMIM 235730
Наша команда профессионалов ответит на ваши вопросы
Синдром Моуат-Вильсона – аутосомно-доминантное заболевание, впервые описано в 1998 году. Клинические характеристики: умственная отсталость, микроцефалия, эпилепсия, низкий рост, врожденные сердечные нарушения, отсроченное моторное развитие. Для больных характерен типичный лицевой фенотип: гипертелоризм (увеличенное расстояние между внутренними краями глазниц), сходящееся косоглазие, широкая носовая перегородка, закругленный кончик носа, постоянно открытый рот, выступающий заостренный подбородок, повернутая назад ушная раковина. При магнитно-резонансном исследовании головного мозга часто обнаруживается гипоплазия или агенезия мозолистого тела (corpus callosum).
В 41-71% случаев у пациентов с синдромом Моуат-Вильсона обнаруживаются признаки болезни Гиршпрунга: отсутствие отхождения мекония в течение первых 24-48 часов жизни, метеоризм, увеличение окружности живота, боли в животе, стойкие запоры, рвота. Как правило, участок аганглиоза у данных больных небольшой. Синдром Моуат-Вильсона, наряду с другими заболеваниями, является несиндромальной формой болезни Гиршпрунга.
В 2001 году описан ген ZEB2 (ZFHX1B, SMADIP1), ответственный за развитие синдрома Моуат-Вильсона. Мутации в гене происходят de novo, как правило, это нонсенс мутации (приводящие к появлению преждевременного стоп-кодона) и мутации со сдвигом рамки считывания. Часто у больных обнаруживаются крупные делеции (от 0.2 до 10.4 млн.пар нуклеотидов) в области 2q22-q24.1, полностью или частично затрагивающие ген ZEB2.
В Центре Молекулярной генетики проводится поиск мутаций в гене ZEB2 методом автоматического секвенирования, а также цитогенетическое исследование для поиска крупных делеций.
Публикации в СМИ
Синдром Картагенера
Синдром Картагенера (триада Картагенера, синдром Зиверта) — транспозиция внутренних органов в сочетании с бронхоэктазами и хроническим синуситом. Дефекты ресничек и жгутиков проявляются при синдроме «неподвижных ресничек» (см. Синдромы разные), возможно развитие рецидивирующего хронического бронхита и синусита. Более половины больных с подобным синдромом имеет situs viscerus inversus — транспозицию внутренних органов (сердце справа, печень слева и т.д.), что в совокупности составляет синдром Картагенера (*244400, 14q32, ген DNECL [600112, тяжёлая цепь динеина], r ). Транспозиция внутренних органов (270100, 14q32, ген SIV, r ) наблюдается также при синдроме Ивмарка.
Клиническая характеристика. Бронхоэктазы, хронический кашель, носовые полипы, синусит, аносмия, дефекты роговицы, средний отит, постоянные головные боли, транспозиция внутренних органов, гипогаммаглобулинемия А, пониженная подвижность лейкоцитов. При синдромах Картагенера и «неподвижных ресничек» сперматозоиды не передвигаются, хотя такие мужчины потенциально фертильны. В этих случаях проводят искусственное оплодотворение с последующим введением концептуса в матку. При синдроме Картагенера и синдроме «неподвижных ресничек» женщины фертильны.
Лечение симптоматическое. Основное внимание уделяется противовоспалительной терапии и поддержанию дренажной функции бронхов (постуральный дренаж, массаж грудной клетки, ингаляции муколитических препаратов). Хирургическое восстановление нормального расположения органов грудной клетки.
МКБ-10 • Q87.8 Другие уточнённые синдромы врождённых аномалий, не классифицированные в других рубриках
Код вставки на сайт
Синдром Картагенера
Синдром Картагенера (триада Картагенера, синдром Зиверта) — транспозиция внутренних органов в сочетании с бронхоэктазами и хроническим синуситом. Дефекты ресничек и жгутиков проявляются при синдроме «неподвижных ресничек» (см. Синдромы разные), возможно развитие рецидивирующего хронического бронхита и синусита. Более половины больных с подобным синдромом имеет situs viscerus inversus — транспозицию внутренних органов (сердце справа, печень слева и т.д.), что в совокупности составляет синдром Картагенера (*244400, 14q32, ген DNECL [600112, тяжёлая цепь динеина], r ). Транспозиция внутренних органов (270100, 14q32, ген SIV, r ) наблюдается также при синдроме Ивмарка.
Клиническая характеристика. Бронхоэктазы, хронический кашель, носовые полипы, синусит, аносмия, дефекты роговицы, средний отит, постоянные головные боли, транспозиция внутренних органов, гипогаммаглобулинемия А, пониженная подвижность лейкоцитов. При синдромах Картагенера и «неподвижных ресничек» сперматозоиды не передвигаются, хотя такие мужчины потенциально фертильны. В этих случаях проводят искусственное оплодотворение с последующим введением концептуса в матку. При синдроме Картагенера и синдроме «неподвижных ресничек» женщины фертильны.
Лечение симптоматическое. Основное внимание уделяется противовоспалительной терапии и поддержанию дренажной функции бронхов (постуральный дренаж, массаж грудной клетки, ингаляции муколитических препаратов). Хирургическое восстановление нормального расположения органов грудной клетки.
МКБ-10 • Q87.8 Другие уточнённые синдромы врождённых аномалий, не классифицированные в других рубриках
Синдром Маршалла

Чекалдина Елена Владимировна
оториноларинголог, к.м.н.
Синдром периодической лихорадки, афтозного стоматита, фарингита и шейного лимфаденита (PFAPA синдром, синдром Маршалла, Periodic Fever, Aphthous Stomatitis, Pharyngitis, Adenitis) — сложное генетическое заболевание и один из наиболее распространенных синдромов периодической лихорадки.
Обычно проявляется в детстве, характеризуется периодическими лихорадками с регулярными интервалами от двух до восьми недель (в среднем 4 недели) и стереотипными клиническими признаками фарингита, афтозного стоматита и шейного лимфаденита. Между эпизодами какие-либо симптомы отсутствуют. Со временем приступы становятся менее тяжелыми, менее частыми и менее продолжительными. У большинства пациентов приступы прекращаются к 10 годам.
Клиническая картина
Периодическая лихорадка — отличительный признак синдрома Маршалла. Начинается внезапно, часто сопровождается ознобом. Температура колеблется от 38,5 до 41 ºC в течение 2-7 дней, а затем резко падает до нормы. До лихорадки у ребенка могут проявляться раздражительность, перепады настроения, он может жаловаться на недомогание, боль в горле, во рту появляются афтозные язвы.
Афтозный стоматит — язвы обычно располагаются на внутренней стороне губ или слизистой оболочки щек, возникают примерно у 40-80% пациентов.
Фарингит (иногда с экссудатом — жидким содержимым в миндалинах, реже с язвами на миндалинах) встречается у 65–100% пациентов.
Шейная лимфаденопатия (увеличение лимфоузлов) сопровождает лихорадку у 60–100% пациентов. Шейные лимфоузлы могут быть болезненными при пальпации.
Другие симптомы: боль в животе (40-65%), боль в суставах (11-42%), рвота (18-41%) и головная боль (18-65%). Реже наблюдаются диарея, кашель, насморк и сыпь.
Во взрослом возрасте эпизоды редко имеют одинаковый интервал между приступами, а при самом приступе редко диагностируется фарингит, при этом чаще возникают боль в груди, головная боль, артралгии (боль в суставах), миалгии (боль в мышцах), глазные симптомы и сыпь.
Диагностика
Диагностических лабораторных тестов для установления PFAPA синдрома не существует. Диагноз в первую очередь ставится на основании истории болезни и результатов осмотра.
В посеве из горла (бактериологическое исследование) может выявляться бета-гемолитический стрептококк группы А (БГСА), но на лечение пенициллинами данные пациенты не отвечают. Это позволяет сделать вывод, что они являются носителями БГСА, то есть имеющиеся симптомы не являются проявлением БГСА-тонзиллофарингита.
В острый период в анализах крови выявляется умеренный лейкоцитоз (13,6 ± 4,5 x 10 9 /л), повышение скорости оседания эритроцитов (СОЭ) и С-реактивного белка (СРБ), нейтропения. Все показатели нормализуются в межприступный период.
PFAPA синдром — это диагноз исключения. В первую очередь исключаются другие причины рецидивирующей лихорадки: инфекция, воспалительные заболевания кишечника, лихорадка при лимфоме Ходжкина, циклическая нейтропения и другие.
Для диагностики имеют значения следующие критерии:
Лечение синдрома Маршалла
Учитывая благоприятное естественное течение, лечение необязательно.
Клинический опыт показывает, что жаропонижающие средства, такие как ацетаминофен (парацетамол) и нестероидные противовоспалительные препараты (ибупрофен), неэффективны в борьбе с другими симптомами PFAPA, кроме лихорадки.
Для лечения в острый период назначаются глюкокортикоиды (преднизолон) в дозировке 1-2 мг/кг (максимальная дозировка 60 мг). Основным недостатком такой терапии является возможное сокращение интервала между приступами. Это происходит у 19–50% пациентов. После прекращения терапии глюкокортикоидами частота эпизодов возвращается к исходному уровню.
У некоторых пациентов в качестве профилактики в межприступный период могут применяться циметидин или колхицин.
Тонзиллэктомия также является вариантом лечения для пациентов, которые не реагируют или не переносят медикаментозное лечение (глюкокортикоиды с профилактической терапией или без нее), или у которых тяжесть эпизодов сильно снижает качество жизни. Риски хирургического вмешательства и доброкачественный долгосрочный характер PFAPA всегда следует принимать во внимание при решении о тонзиллэктомии. У большинства пациентов с PFAPA после тонзиллэктомии сохраняется афтозный стоматит, при этом другие симптомы, включая лихорадку, полностью регрессируют.
Как происходит лечение синдрома Маршалла в клинике Рассвет?
Диагностикой и лечением PFAPA синдрома должен заниматься специалист, хорошо знакомый с данным заболеванием. В клинике Рассвет любой педиатр может провести полноценную дифференциальную диагностику для исключения других причин лихорадки. Только после этого врач назначит диагностическую пробу с глюкокортикоидами, подберет оптимальный вариант лечения и профилактики. После установления диагноза пациенту нет необходимости проводить лабораторные исследования при каждом обострении.
В клинике Рассвет оториноларингологи не назначают антибактериальную терапию при БГСА-носительстве, если у пациента есть подозрение на PFAPA синдром. Тонзиллэктомия не предлагается пациентам с легким течением, так как в возрасте до 10 лет миндалины вносят важный вклад в работу иммунной системы. Удаление миндалин рекомендуется только в случаях неэффективности других методов лечения.
Публикации в СМИ
Синдром Рея
Синдром Рея — острая энцефалопатия с отёком мозга и жировой инфильтрацией органов (преимущественно, печени), возникает у ранее здоровых новорождённых, детей и подростков (чаще в возрасте 4–12 лет), часто связан с предшествующей вирусной инфекцией (например, ветряная оспа или грипп А) и приёмом препаратов, содержащих ацетилсалициловую кислоту.
Клиническая картина • Через 6 дней после начала вирусного заболевания (при ветряной оспе — на 4–5 день после появления высыпаний) — тошнота и неукротимая рвота, сопровождающиеся внезапным изменением психического статуса (варьирует от лёгкой заторможённости до глубокой комы и эпизодов дезориентации, психомоторного возбуждения) • Ухудшение состояния больного с угнетением сознания (гипоксическая энцефалопатия): быстрое развитие коматозного состояния с подавлением основных функций жизнеобеспечения (нарушения ритма дыхания, сердечной деятельности, развитие артериальной гипотензии). Темп развития комы прямо определяет возможный прогноз синдрома (начало заболевания с внезапной потери сознания прогностически благоприятнее постепенного, в течение нескольких часов «входа» в кому) • Увеличение печени в 40% случаев, желтуху наблюдают крайне редко • Развитие тромбогеморрагического синдрома (связанного как с ДВС, так и с явлениями печёночной недостаточности) с типичными проявлениями: от необильной петехиальной сыпи и носовых кровотечений до массивных кровотечений, прямо угрожающих жизни больного. Развернутая клиническая картина тромбогеморрагического синдрома всегда прогностически неблагоприятна • Время от момента госпитализации до наступления смерти — 4 дня • Развитие синдрома всегда следует предполагать у ребёнка, симптоматика заболевания у которого представлена внезапной потерей сознания, судорогами и повторной рвотой (необходимо лабораторное обследование для выявления активности АЛТ, ПТИ и содержания глюкозы крови).
Методы исследования • Анализ крови •• Увеличение содержания АСТ и АЛТ более чем в 3 раза •• Нормальное содержание билирубина •• Повышенное содержание аммиака •• Снижение ПТИ •• Снижение концентрации глюкозы в крови •• Повышение давления ликвора без плеоцитоза (8–10 лейкоцитов/мкл) •• Смешанный дыхательный алкалоз и метаболический ацидоз •• Повышение содержания аминокислот (глутамина, аланина, лизина) • ЭЭГ
Дифференциальная диагностика • Сепсис • Гипертермия (особенно у детей до 1 года) • Нейроинфекции (энцефалиты, менингиты) • Острый гепатит • Отравления (например, фосфором) • Врождённые аномалии синтеза мочевины или окисления жирных кислот • Врождённые нарушения углеводного обмена • Аминоацидурии.
ЛЕЧЕНИЕ
Диета. Исключается пероральное питание.
Тактика ведения • Экстренная госпитализация • Постоянное наблюдение за состоянием ЦНС, ССС, дыхательной системы, водно-электролитным балансом • Контроль за ВЧД (экстренная дегидратация диуретиками с целью декомпрессии мозга) • Катетеризация артерий для исследования газового состава крови и её рН, АД • Интубация трахеи и ИВЛ в режиме гипервентиляции.
Лекарственная терапия • Маннитол 0,5–1 г/кг в/в в возрастной дозировке • Введение в/в жидкости и р-ров электролитов, содержащих 5–10% (иногда до 50%) глюкозы • Антибиотики, не всасывающиеся в ЖКТ, например неомицин 100 мг/кг/сут внутрь в 4 приёма • При выраженном тромбогеморрагическом синдроме — инфузии свежезамороженной плазмы, ингибиторы фибринолиза.
Осложнения Основным осложнением синдрома Рея является вклинение вещества мозга в костные образования черепа (большое затылочное отверстие, вырезка намета мозжечка) с остановкой дыхания и сердечной деятельности • Массивные кровотечения.
Прогноз. Летальность — до 80%. У выживших, длительное время пребывавших в коме возможно развитие резидуальных явлений со стороны ЦНС. Рецидивы редки.
Профилактика. Следует избегать назначения салицилатов при лечении детей с вирусными заболеваниями.
Синоним. Белая печёночная болезнь
МКБ-10 • G93.7 Синдром Рейе
Код вставки на сайт
Синдром Рея
Синдром Рея — острая энцефалопатия с отёком мозга и жировой инфильтрацией органов (преимущественно, печени), возникает у ранее здоровых новорождённых, детей и подростков (чаще в возрасте 4–12 лет), часто связан с предшествующей вирусной инфекцией (например, ветряная оспа или грипп А) и приёмом препаратов, содержащих ацетилсалициловую кислоту.
Клиническая картина • Через 6 дней после начала вирусного заболевания (при ветряной оспе — на 4–5 день после появления высыпаний) — тошнота и неукротимая рвота, сопровождающиеся внезапным изменением психического статуса (варьирует от лёгкой заторможённости до глубокой комы и эпизодов дезориентации, психомоторного возбуждения) • Ухудшение состояния больного с угнетением сознания (гипоксическая энцефалопатия): быстрое развитие коматозного состояния с подавлением основных функций жизнеобеспечения (нарушения ритма дыхания, сердечной деятельности, развитие артериальной гипотензии). Темп развития комы прямо определяет возможный прогноз синдрома (начало заболевания с внезапной потери сознания прогностически благоприятнее постепенного, в течение нескольких часов «входа» в кому) • Увеличение печени в 40% случаев, желтуху наблюдают крайне редко • Развитие тромбогеморрагического синдрома (связанного как с ДВС, так и с явлениями печёночной недостаточности) с типичными проявлениями: от необильной петехиальной сыпи и носовых кровотечений до массивных кровотечений, прямо угрожающих жизни больного. Развернутая клиническая картина тромбогеморрагического синдрома всегда прогностически неблагоприятна • Время от момента госпитализации до наступления смерти — 4 дня • Развитие синдрома всегда следует предполагать у ребёнка, симптоматика заболевания у которого представлена внезапной потерей сознания, судорогами и повторной рвотой (необходимо лабораторное обследование для выявления активности АЛТ, ПТИ и содержания глюкозы крови).
Методы исследования • Анализ крови •• Увеличение содержания АСТ и АЛТ более чем в 3 раза •• Нормальное содержание билирубина •• Повышенное содержание аммиака •• Снижение ПТИ •• Снижение концентрации глюкозы в крови •• Повышение давления ликвора без плеоцитоза (8–10 лейкоцитов/мкл) •• Смешанный дыхательный алкалоз и метаболический ацидоз •• Повышение содержания аминокислот (глутамина, аланина, лизина) • ЭЭГ
Дифференциальная диагностика • Сепсис • Гипертермия (особенно у детей до 1 года) • Нейроинфекции (энцефалиты, менингиты) • Острый гепатит • Отравления (например, фосфором) • Врождённые аномалии синтеза мочевины или окисления жирных кислот • Врождённые нарушения углеводного обмена • Аминоацидурии.
ЛЕЧЕНИЕ
Диета. Исключается пероральное питание.
Тактика ведения • Экстренная госпитализация • Постоянное наблюдение за состоянием ЦНС, ССС, дыхательной системы, водно-электролитным балансом • Контроль за ВЧД (экстренная дегидратация диуретиками с целью декомпрессии мозга) • Катетеризация артерий для исследования газового состава крови и её рН, АД • Интубация трахеи и ИВЛ в режиме гипервентиляции.
Лекарственная терапия • Маннитол 0,5–1 г/кг в/в в возрастной дозировке • Введение в/в жидкости и р-ров электролитов, содержащих 5–10% (иногда до 50%) глюкозы • Антибиотики, не всасывающиеся в ЖКТ, например неомицин 100 мг/кг/сут внутрь в 4 приёма • При выраженном тромбогеморрагическом синдроме — инфузии свежезамороженной плазмы, ингибиторы фибринолиза.
Осложнения Основным осложнением синдрома Рея является вклинение вещества мозга в костные образования черепа (большое затылочное отверстие, вырезка намета мозжечка) с остановкой дыхания и сердечной деятельности • Массивные кровотечения.
Прогноз. Летальность — до 80%. У выживших, длительное время пребывавших в коме возможно развитие резидуальных явлений со стороны ЦНС. Рецидивы редки.
Профилактика. Следует избегать назначения салицилатов при лечении детей с вирусными заболеваниями.
Синоним. Белая печёночная болезнь
МКБ-10 • G93.7 Синдром Рейе
Случай ранней пренатальной диагностики синдрома Тричера Коллинза (Treacher Collins syndrome, OMIM: 154500) 1-й тип, семейная форма


УЗИ сканер RS80
Эталон новых стандартов! Беспрецедентная четкость, разрешение, сверхбыстрая обработка данных, а также исчерпывающий набор современных ультразвуковых технологий для решения самых сложных задач диагностики.
Синдром Тричера Коллинза (СТК) – это врожденное, наследственно обусловленное нарушение развития производных первой глоточной дуги, которое характеризуется специфическими черепно-лицевыми проявлениями: двусторонней симметричной отонижнечелюстной дисплазией с гипоплазией скуловых костей.
Синонимы: синдром Франческетти, синдром Тричера Коллинза–Франческетти, синдром Франческетти–Цвалена–Клейна, челюстно-лицевой дизостоз.
Отличительные признаки СТК: гипоплазия скуловых костей, антимонголоидный разрез глаз, опущенные уголки глаз, колобома нижнего века, пороки развития наружного уха [1, 2].
Хотя первым болезнь описал Аллен Томсон еще в 1846 г., синдром обычно называют именем врача Тричера Коллинза, который в 1900 г. описал двух больных с похожими симптомами. Не верно писать этот синдром через дефис, так как Тричер – имя доктора Коллинза. Уже в 40-х годах прошлого века Адольф Франческетти и Давид Клейн дали подробную характеристику болезни и назвали ее челюстно-лицевым дизостозом [3]. В некоторых странах Европы этот синдром называют синдромом Франческетти или синдромом Тричера Коллинза–Франческетти [4, 5].
Популяционная частота СТК оценивается как 1:50 000 живорожденных [1, 2], однако некоторые авторы называют более частую встречаемость этого синдрома: 1:10 000 [6]. Больные легко узнаваемы, их можно нередко встретить на улицах, увидеть в социальных сетях и, иногда, на телеэкранах. В 2017 г. вышла кинокартина режиссера Стивена Чбоски с Джулией Робертс в главной роли, которая называется «Чудо», где рассказана история мальчика Огги Пулмана с синдромом Тричера Коллинза и прекрасно продемонстрирована вся сложность социальной адаптации таких детей.
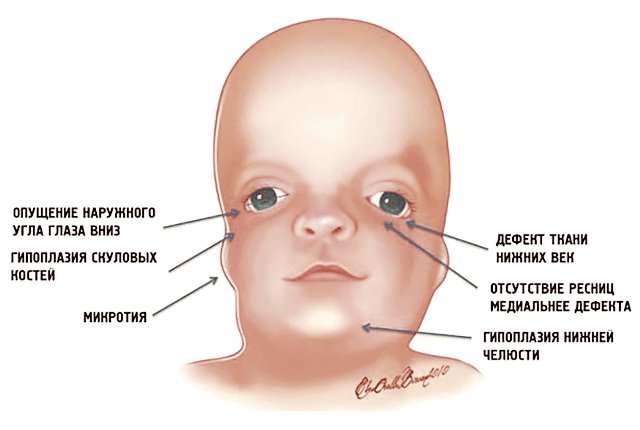
Рис. 1. Схема специфических признаков лицевых дизморфий при синдроме Тричера Коллинза.
Наиболее частые симптомы и фенотипические проявления СТК
У людей с СТК отмечается характерный лицевой дизморфизм (рис. 1) с двусторонней симметричной гипоплазией скуловых костей (95%), характерна гипоплазия инфраорбитального края глазницы (80%) с формированием антимонголоидного разреза глаз (89%) и гипоплазией нижней челюсти (78%), что приводит к аномалии прикуса [1–6], также наблюдается апертогнатия (так называемый открытый прикус). Описана атрезия хоан [7], колобома (расщелина) нижних век между внешней и средней третью (69%), сопровождающаяся отсутствием ресниц. Гипоплазия мягких тканей преимущественно отмечается в скуловой области, нижнем орбитальном крае и щеках. К особенностям относятся сложные нарушения в строении височно-нижнечелюстного сустава, что приводит к ограниченной воз можности открытия рта различной степени тяжести [1].
Часто отмечается аномалия наружного уха, например микротия или анотия (77%), атрезия наружного слухового прохода и аномалии развития слуховых костей (60%), что приводит к кондуктивной тугоухости [1–8]. Снижение зрения, вплоть до полной его потери, встречается в 37% случаев. Нёбо высокое, имеет готическую форму и иногда наблюдается его расщелина (28%).
Умственные способности, как правило, нормальные. Умственная отсталость встречается лишь у 5% людей с СТК [1, 2]. Из-за узких верхних дыхательных путей и ограниченного открывания рта в раннем возрасте могут возникать трудности с дыханием и питанием [8]. Из частых признаков описан чрезмерный рост волос на щеках [2, 8, 9].
Этиология синдрома Тричера Коллинза
На сегодняшний день описано три типа СТК. До 93% всех случаев – это синдром 1-го типа [10]. СТК 1-го типа связан с мутациями гена TCOF1, который расположен в сегменте 5q32 – q33. Тип наследования аутосомно-доминантный [2] с 90% пенетрантностью и переменной экспрессивностью (проявляемостью), даже у пациентов в пределах одной семьи. Известны наблюдения детей с выраженными клиническими проявлениями синдрома в одной семье, тогда как у одного из их родителей была обнаружена та же мутация без выраженных клинических проявлений болезни [2, 4–6]. Около 60% случаев СТК не наследуются от больных родителей, а являются новыми мутациями (de novo).
Также описаны 2-й и 3-й типы СТК. Второй тип вызван мутацией гена POLR1D на хромосоме 13q12, 3-й тип – мутацией гена POLR1C на хромосоме 6p21. Нужно отметить, что клинически все три типа не отличаются друг от друга, несмотря на то что мутации затрагивают разные гены, на разных хромосомах [2] и тип наследования может быть и аутосомно-рецессивным [11].
Пренатальная диагностика СТК
Несмотря на давно описанный в литературе и хорошо известный врачам-генетикам диагноз, количество статей, посвященных случаям дородовой диагностики СТК, весьма ограничено. Это связано с трудностью визуализации и объективизации некоторых классических фенотипических признаков синдрома при проведении пренатальной эхографии [12]. Ультразвуковые проявления изменений лицевого фенотипа у плодов бывают не очевидны, и часто рождение таких детей является полной неожиданностью не только для их родителей, но и для врачей пренатальной диагностики. Явные после рождения «ядерные» признаки СТК, такие как гипоплазия скуловых костей, микрогнатия, расщелина нёба, колобома нижнего века, антимонголоидный разрез глаз, отсутствие ресниц, чаще всего остаются незамеченными, даже при современных возможностях ультразвуковых приборов, особенно когда нет генетической настороженности при осмотре, что бывает при возникновении мутации de novo у фенотипически здоровых родителей. Часто в пренатальном периоде могут наблюдаться многоводие и задержка роста плода [14, 15]. Внедрение в клиническую практику современных режимов сканирования при помощи объемной визуализации лицевого фенотипа значимо облегчает диагностику [16]. Положение глазных щелей, аномальная форма носа, низко расположенные уши – все эти хорошо известные основные признаки СТК очень сложно уверенно визуализировать в обычном рутинном 2D-режиме, но при применении 3D-технологий их дефиниция становится более очевидной [16, 17].
Дифференциальная диагностика СТК должна включать некоторые генетические синдромы с преимущественным поражением лицевых структур [17]:
Следует подчеркнуть, что аномалии конечностей не свойственны для СТК и для синдрома Пьера Робена, и, если они присутствуют, следует больше думать о синдромах Миллера или Нагера.
Профилактика и лечение СТК
Генетическое консультирование семей с больным ребенком/плодом осложняется вариабельной проявляемостью заболевания и должно осуществляться мультидисциплинарной группой специалистов по пренатальной диагностике с обязательным выяснением этиологии возникновения заболевания в конкретной ситуации (семейная форма либо мутация de novo). При наличии у родителя признаков СКР единственным эффективным методом профилактики заболевания следует назвать применение методик экстракорпорального оплодотворения с предимплантационной диагностикой с целью переноса здоровых эмбрионов, либо применение донорских ооцитов или сперматозоидов.
При продолжающейся беременности послеродовое ведение требует междисциплинарного подхода (акушер, неонатолог, хирург, анестезиолог и генетик); и из-за возможных острых проблем с дыханием роды должны планироваться в специализированных перинатальных центрах. Лечение больных с СТК многопрофильное. В случае возникновения постнатального респираторного дистресс-синдрома необходимо применение трахеостомии, неинвазивной вентиляции и дистракции нижней челюсти. Челюстно-лицевая и пластическая хирургия позволяет устранить гипоплазию мягких тканей (коррекция овала лица с помощью липоскульптуры), гипоплазию костной ткани (хирургическая дистракция кости, костные трансплантаты), колобому век и расщелину нёба (хирургическое восстановление). Для устранения аномалий среднего уха (функциональная хирургия) и наружного уха (реконструкция ушных раковин) требуется участие специалиста в области ЛОР-хирургии. Коррекция нарушения слуха должна осуществляться на ранней стадии (слуховые аппараты и функциональная хирургия), что способствует нормальному развитию ребенка.
При надлежащем лечении прогноз для легких форм заболевания является благоприятным. Для тяжелых форм заболевания с выраженными клиническими проявлениями прогноз неблагоприятный не только для здоровья, но и для жизни.
Описание случая синдрома Тричера Коллинза
В медико-генетическом отделении (МГО) Московского областного НИИ акушерства и гинекологии для консультации по прогнозу потомства и возможностях обследования обратилась пациентка 25 лет со сроком беременности 8 нед. Данная беременность вторая. Брак не родственный. Муж здоров, производственных вредностей супруги не имеют. Первая беременность закончилась преждевременными родами в сроке 36 нед. Родилась девочка с массой тела 1990 г, ростом 51 см, с оценкой по шкале Апгар 7/7 баллов. При осмотре ребенка генетиком выявлены особенности фенотипа, характерные для СТК: гипоплазия скуловых костей, антимонголоидный разрез глаз, гипоплазия нижней челюсти, двусторонняя микротия с атрезией слуховых проходов. Методом автоматического прямого секвенирования был проведен поиск мутаций в гене TCOF1. Выявлен патогенный вариант c.3946_3947 delGA в гетерозиготном состоянии. Ребенку выставлен клинический диагноз: синдром Тричера Коллинза. Тяжесть состояния ребенка усугубилась врожденной пневмонией, церебральной ишемией II степени, недоношенностью, анемией тяжелой степени. Ребенок был переведен в отделение реанимации, умер в 1,5 мес. При консультировании ребенка генетиком риск повторного рождения больного ребенка в семье расценен как низкий, так как данная мутация расценена генетиком как мутация de novo. Дана рекомендация о пренатальной диагностике и кариотипировании плода при следующей беременности без указания на необходимость специфической диагностики СТК. Пациентка самостоятельно обратилась для обследования в медико-генетический научный центр (МГНЦ). В образце ее ДНК методом прямого автоматического секвенирования была найдена патогенная мутация в гене TCOF1 в гетерозиготном состоянии. Таким образом, у пациентки тоже имеется СТК и риск рождения у нее больных детей будет высоким – 50%. При предыдущем осмотре генетиком ее фенотип был не изучен и не оценен в полном объеме. При внимательном осмотре пациентки найдены мягкие, но классические признаки СТК: опущенные уголки глаз, колобомы нижнего века, рост волос на лице, гипоплазия мягких тканей в области скуловых дуг. При сборе анамнеза выяснено, что пациентка страдает двусторонней тугоухостью. С учетом аутосомно-доминантного типа наследования СТК, известного картированного патологического гена было рекомендовано проведение инвазивной пренатальной диагностики с прицельным поиском известной мутации и экспертное ультразвуковое исследование в 12–13 нед беременности.
При ультразвуковом исследовании выявлены множественные особенности лицевого фенотипа у плода: микрогнатия (рис. 2–4), треугольная форма лица (рис. 5), опущенные книзу глазницы и гипоплазия скуловых дуг (рис. 6, 7), аномальная форма и положение ушей (рис. 5, 7).