Синдром стиклера что это
Штиклер с сотр. (Stickler el al.) впервые определили как синдром состояние, заключающееся в прогрессирующей множественной эпифизарной дисплазии и других скелетных аномалиях, разболтанности суставов, близорукости, отслойке сетчатки, расщеплении: неба и глухоте. Ранее случай заболевания был описан David.
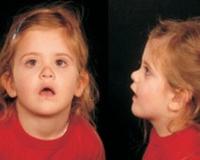
Клинические данные. Лицо. Почти у 50% больных отмечается уплощение средней части лица. Нос часто седловидный (David, Spranger, Falger et al.). Stickler и Pugh (1967), Spranger, Falger с сотр. и многие другие исследователи (см. «Диагноз») отметили расщепление неба, вероятно, в 50% случаев.
Костно-мышечная система. В течение нескольких первых лет жизни отмечается утолщение костей лодыжек, коленей и кистей. Суставы какое-то время болезненны при движениях, а затем развиваются анкилоз и полная неподвижность. Изредка суставы гиперемированы и горячи на ощупь. Больные испытывают трудности при ходьбе и быстро устают. Примерно в 30% случаев наблюдается кифоз грудного отдела позвоночника, благодаря чему создается впечатление чрезмерной длины рук (David, Stickler et al.).
Другие аномалии скелета, такие, как килевидная грудь, вальгусная деформация коленей, разболтанность суставов, конская стопа и плоскостопие, встречаются примерно в 60% случаев.
Орган зрения. Почти у 70% больных была обнаружена врожденная прогрессирующая близорукость, достигавшая 18 диоптрий. Почти у 20% из них до десятилетнего возраста выявлялась широкая зона отслойки сетчатки. У 10% больных встречаются вторичная катаракта, кератопатия и/или глаукома.

Орган слуха. Stickler и Pugh описали нейросенсорную глухоту в пределах 25—30 дБ. Spranger сообщил о проводящей глухоте. David не охарактеризовал глухоту, которую обнаружил у своих больных. Наши личные наблюдения позволяют предположить, что нейросенсорная или смешанная глухота встречается примерно у 15% больных. Такая же частота была установлена Herrmann (личное сообщение). Popkin и Polomeno сообщили о двух больших семьях, в которых примерно у 10% больных была обнаружена нейросенсорная глухота.
Вестибулярная система. Результаты исследований не опубликованы.
Лабораторные данные. Рентгенологически почти у 1/3 больных отмечаются множественные нарушения окостенения эпифизов и уменьшение ширины диафизов длинных костей. Истонченные диафизы контрастируют с метафизами, имеющими нормальную ширину. Кости таза гипопластичны, а головка бедра плохо сформирована, округлой формы, и бедро находится в вальгусном положении. Тела позвонков уплощены,. неправильной формы, с тенденцией к клиновидной деформации спереди. Постоянно наблюдается кифоз в грудном отделе позвоночника.
Наследственность. Многочисленные родословные подтверждают аутосомно-доминантное наследование.
Диагноз. Cohen с соавт. описали проблему отграничения синдрома Штиклера от заболевания, которое называется синдромом Червенка (Cervenka) и заключается в уплощении средней части лица, расщеплении неба, близорукости, отслойке сетчатки и аутосомно-доминантном наследовании (Frandsen, Smith, van Balen, Falger, Hirose et al.). Hall) убедительно показал, что синдром Червепка представляет собой легкую форму наследственной артро-офтальмопатии. Семья, описанная Schreiner с сотр., продемонстрировала выраженную иариабелыюсть экспрессивности синдрома Штиклера.
Данные, представленные Kozlowski с соавт., были недостаточно хорошо документированы, для того чтобы различить, имеется ли у их больных синдром Штиклера или другой синдром, включающий скелетные аномалии и глухоту.
Лечение. Показано раннее направление к офтальмологу.
Прогноз. Артропатия прогрессивно ухудшается. Отслойка сетчатки рецидивирует, вопреки хирургическому лечению.
Выводы. Характерным для этого синдрома является: 1) аутосомно-доминантное наследование; 2) многочисленные, по часто слабо выраженные нарушения окостенения, включающие патологию эпифизов, сужение диафизов и платиспондилию; 3) разболтанность суставов; 4) гипоплазия средней части лица; 5) резкая миопия и часто отслойка сетчатки; 6) иногда расщепление неба, подслизистое расщепление неба или раздвоение язычка; 7) смешанная глухота.
Синдром Стиклера ( Врожденная артроофтальмопатия )

Синдром Стиклера — это редкий вариант наследственных коллагенопатий, при котором происходит нарушение структуры коллагена II, IX, XI типов. Наследуется по аутосомно-доминантному и аутосомно-рецессивному механизмах. Заболевание проявляется прогрессирующей миопией с отслойкой сетчатки, поражением костно-суставного аппарата, характерными фенотипическими особенностями. Диагностика синдрома проводится при помощи рентгенографии скелета, офтальмоскопии, биохимического и молекулярно-генетического тестирования. Лечение поддерживающее: реабилитационные программы, психологическая поддержка, хирургическая коррекция осложнений.
МКБ-10
Общие сведения
Синдром Стиклера назван в честь американского педиатра Г.Б. Стиклера, который в 1965 г. выпустил статью с анализом своей многолетней работы с пациентами, имевшими типичный комплекс симптомов: прогрессирующую миопию, дегенерацию суставов, расширение эпифизов. Генетические основы патологии были открыты только в 1980-1990-х гг. Заболевание имеет второе название «врожденная артроофтальмопатия». Распространенность синдрома Стиклера составляет 1 случай на 7,5-10 тыс. взрослого населения, половых различий в структуре заболеваемости нет.
Причины
При синдроме Стиклера выделено 5 вариантов мутаций в генах, кодирующих синтез коллагеновых волокон: COL2A1, COL11A1, COL11A2, COL9A1, COL9A2. Заболевание 1, 2, 3 подтипов характеризуется аутосомно-доминантным вариантом наследования, то есть вероятность рождения больного ребенка составляет 50%, если один из родителей — носитель мутантного гена. Синдром 4 и 5 подтипов передается аутосомно-рецессивно, проявляется только если оба родителя имеют мутантный ген.
Патогенез
Болезнь возникает при нарушении структуры коллагеновых волокон 2-го, 9-го и 10-го типов, которые входят в состав костной и хрящевой ткани, стекловидного тела глазного яблока. Дефектный коллаген изменяет нормальную структуру соединительной ткани. При этом у пациентов нарушается формирование суставов, происходят дегенеративные процессы, деформируется костный скелет и, как следствие, становятся заметными характерные фенотипические признаки.
Классификация
В клинической практике систематизация заболевания на отдельные подтипы используется редко, что обусловлено схожестью их симптоматики, общностью подходов лечения. По результатам молекулярно-генетической диагностики были выделены 5 разновидностей синдрома Стиклера, которые отличаются локализацией мутации, видом дефектного коллагена. В классификации наследственной коллагенопатии выделяют такие варианты:
Симптомы
Дети с врожденной артроофтальмопатией имеют специфические изменения черт лица, которые заметны уже в неонатальном периоде. У больных проявляется аномалия Робина: плоское лицо с недоразвитой нижней челюстью, расщелины неба, западение языка. Также наблюдается выпирание верхней губы, деформация скуловых дуг, западение переносицы. Изредка пациенты имеют типичный марфаноидный фенотип, что затрудняет постановку диагноза.
Суставные поражения вначале проявляются гипермобильностью, чрезмерной гибкостью и подвижностью. Дети и подростки часто страдают искривлениями позвоночника в виде кифоза, лордоза, сколиоза. По мере прогрессирования синдрома дегенеративные изменения провоцируют контрактуры, боль, увеличение и припухлость суставов. Пациенты после 30 лет обычно сталкиваются с артропатией тазобедренных, коленных, голеностопных суставов.
Повреждение зрительного аппарата состоят в прогрессирующем ухудшении зрения, появлении катаракты, дегенеративных изменений стекловидного тела. Возникают нарушения слуха по типу кондуктивной тугоухости. У части больных отмечается задержка психомоторного развития, снижение интеллектуальных способностей легкой или средней степени тяжести.
Осложнения
Наиболее опасным последствием синдрома является отслойка сетчатки, которая может начаться в детском возрасте при отсутствии лечения прогрессирующей миопии. В результате наступает слепота на один или оба глаза. Поражение суставов со временем приводит к ограничению подвижности и без коррекции заканчивается инвалидностью больных. Неэстетичные изменения лицевого скелета становятся причинами комплексов, проблем с социализацией.
Вовлечение в процесс слухового анализатора чревато частыми отитами, а при неблагоприятном течении есть риск снижения слуха вплоть до глухоты. Изредка патология соединительной ткани затрагивает клапанный аппарат сердца, что вызывает пролапс митрального клапана, нарушения кровообращения, сердечную недостаточность. Аномалии строения ротовой полости сопровождаются проблемами с употреблением пищи, высоким риском аспирации, затруднениями при дыхании.
Диагностика
Синдром Стиклера отличается полиморфностью клинической картины, схожестью симптоматики с другими наследственными нарушениями соединительной ткани, что усложняет диагностический поиск. При первичном осмотре у педиатра определяются фенотипические проявления коллагенопатий, изучается семейный анамнез. Для подтверждения врожденной артроофтальмопатии назначаются следующие методы исследования:
Лечение синдрома Стиклера
Консервативная терапия
Этиопатогенетическое лечение болезни Стиклера отсутствует. Усилия врачей направлены на коррекцию существующих проблем со здоровьем, повышение качества жизни пациентов. Комплексный подход к терапии таких больных включает несколько направлений:
Хирургическое лечение
Больным зачастую требуется помощь офтальмохирургов, которые проводят лазерную коагуляцию или криотерапию для лечения отслоения сетчатки, сохранения зрения. При критическом ухудшении слуха рассматривается вопрос о постановке слухового аппарата. Аномалии развития лицевого черепа требуют поэтапной пластической коррекции, чтобы устранить проблемы с дыханием и приемом пищи, улучшить эстетику внешности.
Прогноз и профилактика
Состояние пациентов зависит от своевременности постановки диагноза, полноты терапевтических, хирургических, реабилитационных мероприятий. При должном лечении удается поддерживать хорошее качество жизни больных, поэтому прогноз относительно благоприятный. Меры первичной профилактики синдрома не разработаны. Семейным парам с отягощенной наследственностью рекомендована консультация генетика при планировании зачатия.
Синдром стиклера что это
Наследственные витреоретинопатии вызывают большинство случаев отслоек сетчатки у детей; Meredith и Snead предлагают классифицировать их следующим образом;
1. Витреоретинопатии, сопутствующие аномалиям скелета.
2. Витреоретинопатии, сопровождающиеся прогрессирующей дисфункцией сетчатки.
3. Витреоретинопатии, сопровождающиеся аномальной сосудистой сетью сетчатки.
4. Витреоретинопатии, сопровождающиеся аномалиями роговицы.
При этой патологии отслойки обычно тяжелые и часто связаны с гигантскими разрывами.
Витреоретинопатии, сопутствующие скелетным аномалиям, составляют самую большую подгруппу заболеваний. Из них большинство случаев вызывается различными вариантами синдрома Стиклера.

а) Синдромы Стиклера. Синдромы Стиклера составляют часть спектра коллагенопатий II/XI типа, включающего в себя также дисплазию Книста (MIM 156550) и врожденную спондилоэпифизарную дисплазию (spondyloepiphyseal dysplasia congenita — SEDC, MIM 183900). В настоящее время идентифицировано по меньшей мере шесть клинически различных подгрупп, генетическую гетерогенность еще предстоит изучить.
Большинство случаев, с которыми сталкиваются офтальмологи, относятся к синдрому Стиклера I типа, наследуемому по аутосомно-доминантному типу. Однако значительная меньшая часть вызывается возникшими de novo мутациями, поэтому семейный анамнез не имеет диагностической значимости. Более того, существуют «только глазные» варианты, при которых системные проявления слишком малочисленны, чтобы заподозрить диагноз — клиницист, будь бдителен!
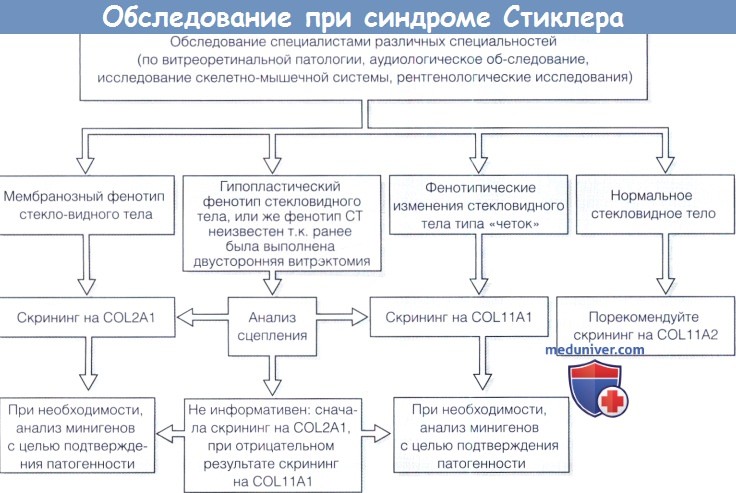
1. Диагностика. До выполнения стандартного молекулярного генетического анализа диагноз полностью основывается на комбинации больших и малых клинических критериев. Тем не менее, исследование клинического фенотипа значительно помогает определить направление молекулярного генетического анализа в соответствии с алгоритмом, приведенным на рисунке ниже.
2. Клинические проявления. Изменения глаз. Основной признак синдрома Стиклера — врожденная аномалия развития стекловидного тела, проявляющаяся видимым при осмотре на щелевой лампе нарушением его архитектоники. Этот патогномоничный признак имеет большое значение для клинической диагностики и дифференцировки подтипов заболевания, не сопровождающихся системной патологией и проявляющихся только изменениями глаз.
Большинство таких пациентов, попавших на прием к офтальмологу, будут иметь синдром Стиклера первого или второго типа, часто у них наблюдается близорукость. Аутосомно-рецессивный синдром Стиклера, развивающийся вследствие мутаций гена коллагена IX типа, встречается редко.
Отмечаются врожденные аномалии рефракции высокой степени, часто они сопровождаются выраженным астигматизмом. В некоторых сериях наблюдений аномалии рефракции отсутствовали почти у четверти пациентов. Точно установлена связь с врожденными катарактами, у некоторых пациентов наблюдается характерное квадрантное ламеллярное кортикальное помутнение хрусталика, что может являться информативным диагностическим признаком. Однако он не помогает дифференцировке различных подгрупп, поскольку наблюдается при синдроме Стиклера и первого, и второго типа.
Риск отслойки сетчатки среди пациентов с клиническим диагнозом одного из типов синдрома Стиклера в Великобритании и США превышает 50%. В подгруппе пациентов с генетически подтвержденным синдромом Стиклера 1 типа эта цифра превышает 70%, из которых почти у половины развивается двусторонняя отслойка. Риск отслойки при синдроме Стиклера 2 типа не рассчитан, но он несколько меньше, вероятно, между 40% и 50%.
У пациентов с синдромом Стиклера 1 типа отмечается предрасположенность к развитию отслойки вследствие гигантского разрыва сетчатки, определяемого как круговой разрыв у соединения с плоской частью цилиарного тела и развивающегося вследствие того, что зона отслойки задней гиалоидной мембраны (ЗГМ, posterior hyaloid membrane, РНМ) по сравнению с нормой значительно смещена вперед. В таких условиях (при наличии гигантского разрыва сетчатки или без него), задняя гиалоидная мембрана оказывается фиксированной сзади от уже существующей аномалии 1 типа, при этом наблюдается характерная картина двойной мембраны.
Клиницист должен проявлять настороженность в отношении отслойки заднего витреума и при синдроме Стиклера 2 типа, когда у ребенка визуализируется (одна) отслоенная задняя гиалоидная мембрана, симулирующая аномалию 1 типа. При синдроме Стиклера 1 типа профилактическая ретинопексия, выполняемая и локализуемая с целью профилактики прогрессирования гигантского разрыва сетчатки, значительно снижает риск отслойки сетчатки и слепоты вследствие этого вида разрыва. Может развиться непредсказуемая отслойка вследствие более центральных разрывов, в такой ситуации профилактическая ретинопексия неэффективна.
Системные проявления. При всех подгруппах синдрома Стиклера наблюдаются расщелины твердого или мягкого неба, отличающиеся от расщелин средней линии другой этиологии, часто сопровождающихся расщелинами губы. У пациентов с синдромом Стиклера расщелина губы обычно отсутствует. Аномалии твердого и мягкого неба часто сопровождаются дисфункцией евстахиевой трубы и поражением среднего уха. Если оперативное восстановление неба не выполнялось, выявить субклинические расщелины можно при прямом осмотре и пальпации —это еще один часто упускаемый диагностический признак.
Расщелины средней линии часто сопровождаются нарушениями слуха, но у многих пациентов выявляется скрытое субклиническое ухудшение восприятия высоких частот — сенсоневральный дефект вследствие аномалий улитки.
Более чем у 80% детей с синдромом Стиклера описываются мышечно-скелетные аномалии, которые могут значительно ухудшать качество жизни. У некоторых детей наблюдается увеличение суставов вследствие эпифизарной дисплазии. Часто отмечается гипермобильность суставов и, хотя она может не сопровождаться жалобами, приводит к Х-образноп деформации коленных суставов, плоскостопию, подвывихам или вывихам суставов и обширным суставным болям. Иногда пациентам ставится ошибочный диагноз болезни Петерса из-за сходной рентгенологической картины. Также подросткам с синдромом Стиклера ставится диагноз болезни Osgood-Schlatter, но, поскольку это в большей степени клинический, а не рентгенологический диагноз (и часто является причиной боли по передней поверхности большеберцовой кости у подростков), характер этого сочетания еще предстоит выяснить.
б) Дисплазия Книста (MIM 156550). Дисплазия Книста — аутосомно-доминантное заболевание, характеризующееся многими общими проявлениями с синдромом Стиклера; вызывающий его ген располагается в том же локусе. Выявляются мутации в том же гене, что и при синдроме Стиклера 1 типа (COL2A1), но они приводят к доминант-негативному эффекту, а не гаплонедостаточности, что сопровождается значительно более тяжелой артропатией. Заболевание обычно проявляется при рождении укорочением туловища и конечностей, врожденным мегалофтальмом и плоской переносицей. Обычно при рождении суставы увеличены, пальцы длинные и узловатые.
Прохождение вех двигательного развития замедлено вследствие деформации суставов, может развиваться дисфункциональная мышечная атрофия. Как и при синдроме Стиклера, может развиваться и кондуктивная, и сенсоневральная тугоухость. Интеллект нормальный, основными офтальмологическими осложнениями являются близорукость, гигантские разрывы и отслойка сетчатки.
в) Врожденная спондилоэпифизарная дисплазия (MIM 183900). Врожденная спондилоэпифизарная дисплазия (spondyloepiphyseal dysplasia congenita — SEDC) проявляется при рождении укорочением туловища и, в меньшей степени, конечностей (ризомелическое укорочение конечностей). Это аутосомно-доминантное заболевание, обычно оно вызывается доминантно-негативными мутациями гена коллагена II типа (COL2A1). Как при других коллагенопатиях II типа, заболевание проявляется врожденными аномалиями развития стекловидного тела, кондуктивной и сенсоневральной тугоухостью и расщелиной неба.
У пациентов развивается бочкообразная грудная клетка и патологический поясничный лордоз, который может нарушать дыхательную функцию. Может наблюдаться гипоплазия второго шейного отростка, предрасполагающая к цервикомедуллярной нестабильности, поэтому перед общей анестезией необходимо выполнить лучевое исследование шейного отдела позвоночника. Так же как и при других коллагенопатиях II/XI типов, у пациентов с врожденной спондилоэпифизарной дисплазией отмечается повышенный риск регматогенной отслойки сетчатки.
г) Синдром Маршала (MIM 154780). Остается неясным, являются ли синдром Маршала и синдром Стиклера 2 типа различными заболеваниями, или нет. У них много общих проявлений, в том числе гипоплазия средней зоны лица, спондилоэпифезарные аномалии, расщелина неба и сенсоневральная тугоухость, у пациентов с синдромом Marshall также наблюдается эктодермальная дисплазия с гипертрихозом и гипогидрозом, утолщением костей свода черепа, гипертелоризм. От термина «синдром Маршала-Стиклера» следует отказаться, пока синдром Маршала не будет лучше изучен.
д) Синдром Кноблоха (MIM 267750). Синдром Кноблоха — аутосомно-рецессивное заболевание, проявляющееся близорукостью высокой степени, нистагмом, врожденной витреоретинопатией, отслойкой сетчатки, врожденным затылочным энцефалоцеле, нетипичными ладонными складками, гипоплазией ногтей и кариесом зубов. При молекулярном генетическом анализе обычно выявляются изменения сплайс-сайта гена, кодирующего а 1-цепь коллагена XVIII типа (COL18A1; 21q22.3; 46kb; 41 экзона), хотя также идентифицированы компаунд-гетерозиготы.
COL18A1 экспрессируется в двух различных изоформах, считается, что более длинная изоформа участвует в поддержании структуры сетчатки, а также в закрытии нервной трубки. Он интенсивно экспрессируется в почках и печени, но у пациентов не описано значительных аномалий этих органов. У COL18A1 нокаут-мышей наблюдаются аномалии увеального тракта.
е) Синдром Марфана (MIM 154700). Синдром Марфана — аутосомно-доминантное заболевание соединительной ткани, сопровождающееся аномальной архитектоникой стекловидного тела, миопическим астигматизмом и характерными изменения скелета, высоким ростом, непропорционально длинными конечностями и пальцами, сколиозом, поясничным лордозом, гиперподвижностью суставов, узким высоким (но без расщелины) небом и деформациями передней части грудной клетки. Причиной этого заболевания является патология фибриллина, высокомолекулярного экстрацеллюлярного гликопротеина. Мутации гена фибриллина хромосомы 15 (FBN1) вызывают как синдром Марфана, так и доминантную эктопию хрусталика. Другие изменения глаз включают в себя отслойку сетчатки, близорукость, мегалофтальм, плоскую роговицу, гипоплазию радужки, глаукому и рано дебютирующую ядерную склеротическую катаракту.
Сообщается, что сопутствующая регматогенная отслойка сетчатки возникает в 8-50% случаев; примерно 75% из них диагностируются в возрасте младше 20 лет. Близорукость обычно развивается с возрастом, в одной большой серии наблюдений не было выявлено ни одного случая миопии в возрасте младше трех лет, в отличие от врожденной непрогрессирующей близорукости, характерной для синдрома Стиклера 1 типа.
При синдроме Марфана вследствие структурных аномалий радужки зрачки расширяются плохо, при этих изменениях с подвывихом хрусталика и рыхлой склерой оперативное лечение отслойки сетчатки становится трудной задачей. Часто требуются ленсэктомия через плоскую часть цилиарного тела и тампонада витреальной полости.
Сердечно-сосудистые аномалии включают в себя пролапс митрального клапана, митральную регургитацию, расширение корня аорты и аортальную регургитацию; основными жизнеугрожающими осложнениями являются аневризма и расслоение аорты.
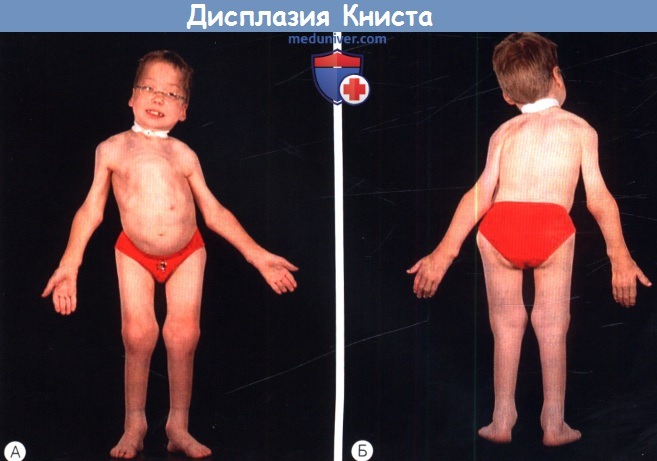 Дисплазия Книста.
Дисплазия Книста.
Обратите внимание на близорукость высокой степени и тяжелую артропатию,
особенно поражение позвоночника, колен, локтей и дисплазию фаланг. 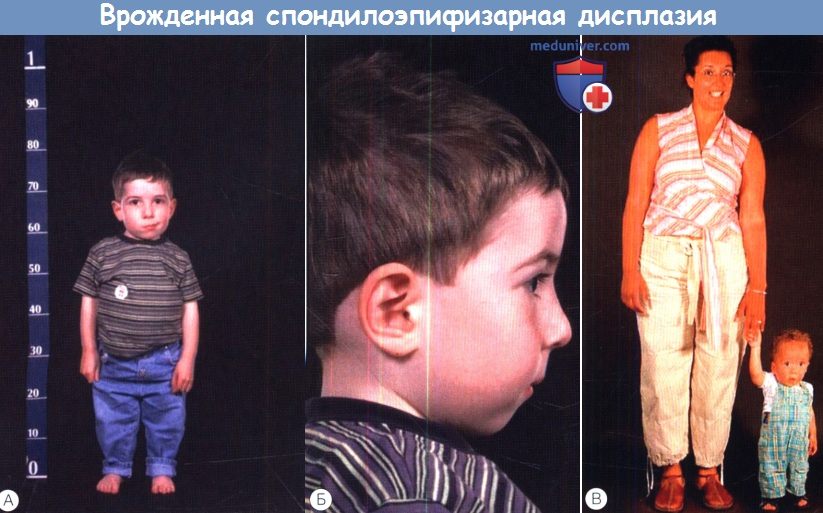 Врожденная спондилоэпифизарная дисплазия.
Врожденная спондилоэпифизарная дисплазия.
Обратите внимание на маленький рост, бочкообразную грудную клетку и ризомелическое (проксимальное) укорочение конечностей.
У пациентов обычно наблюдается аномалия развития — мембранозное стекловидное тело. 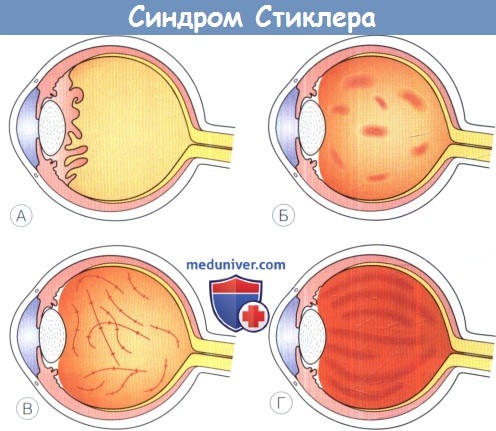 Схематическое изображение фенотипов стекловидного тела при синдроме Стиклера.
Схематическое изображение фенотипов стекловидного тела при синдроме Стиклера.
(А) Врожденная аномалия — мембранозное стекловидное тело. (Б) Врожденные аномалии стекловидного тела типа «четок».
(В) Врожденная аномалия — гипоплазия стекловидного тела. (Г) Нормальная архитектоника: компактная ламеллярная структура.  Классическая квадрантная ламеллярная катаракта, наблюдающаяся при синдроме Стиклера 1 и 2 типов.
Классическая квадрантная ламеллярная катаракта, наблюдающаяся при синдроме Стиклера 1 и 2 типов. 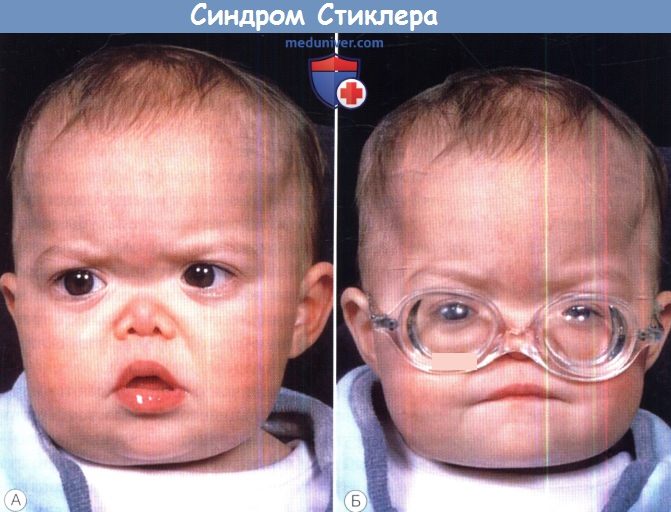 Синдром Стиклера.
Синдром Стиклера.
Выраженная гипоплазия носа и врожденная близорукость, диагностированная при рефрактометрии в условиях циклоплегии. 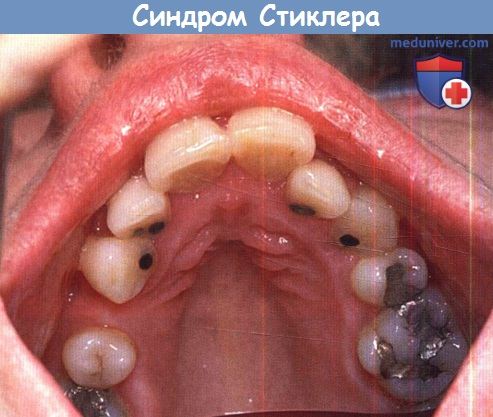 Синдром Стиклера.
Синдром Стиклера.
Даже если в анамнезе отсутствует пластика неба, не забудьте произвести его осмотр.
Этот пациент полагал, что с его небом «все в порядке». 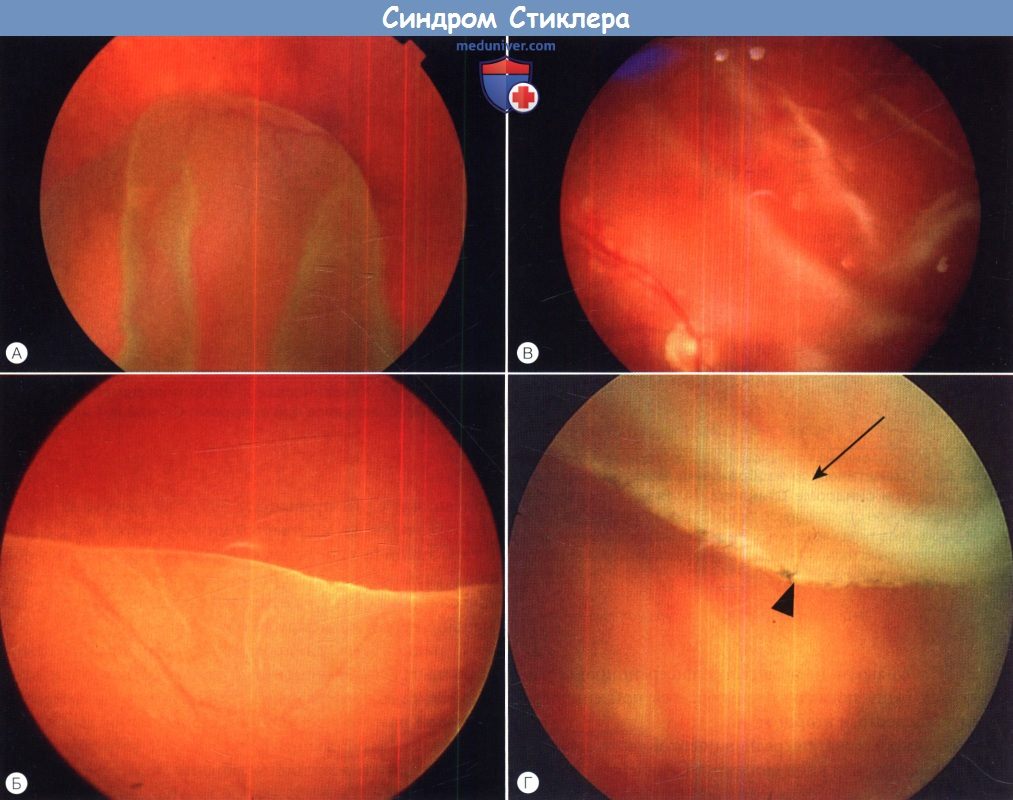 Отслойки при гигантских разрывах сетчатки при синдроме Стиклера 1 типа.
Отслойки при гигантских разрывах сетчатки при синдроме Стиклера 1 типа.
Обратите внимание на профилактические лазерокоагуляты на фото (Г), они были нанесены слишком далеко на периферии, чтобы предотвратить отслойку.
Стрелкой показан гигантский разрыв сетчатки; сплошной треугольник указывает на экваториальные лазерокоагуляты. 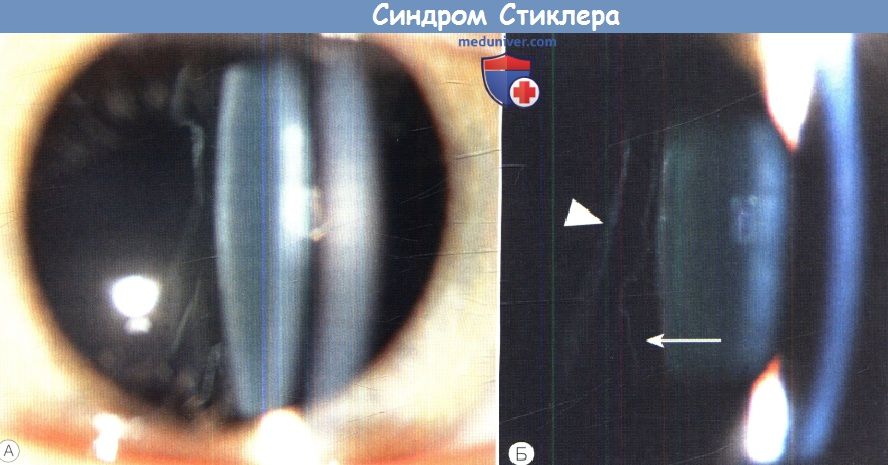 (А) Синдром Стиклера 1 типа—типичная врожденная аномалия — мембранозное стекловидное тело.
(А) Синдром Стиклера 1 типа—типичная врожденная аномалия — мембранозное стекловидное тело.
(Б) Отслойка заднего витреума при синдроме Стиклера 1 типа.
Позади мембранозного стекловидного тела — врожденной аномалии 1 типа (стрелка) видна задняя гиалоидная мембрана (треугольник), что создает картину двойной мембраны. 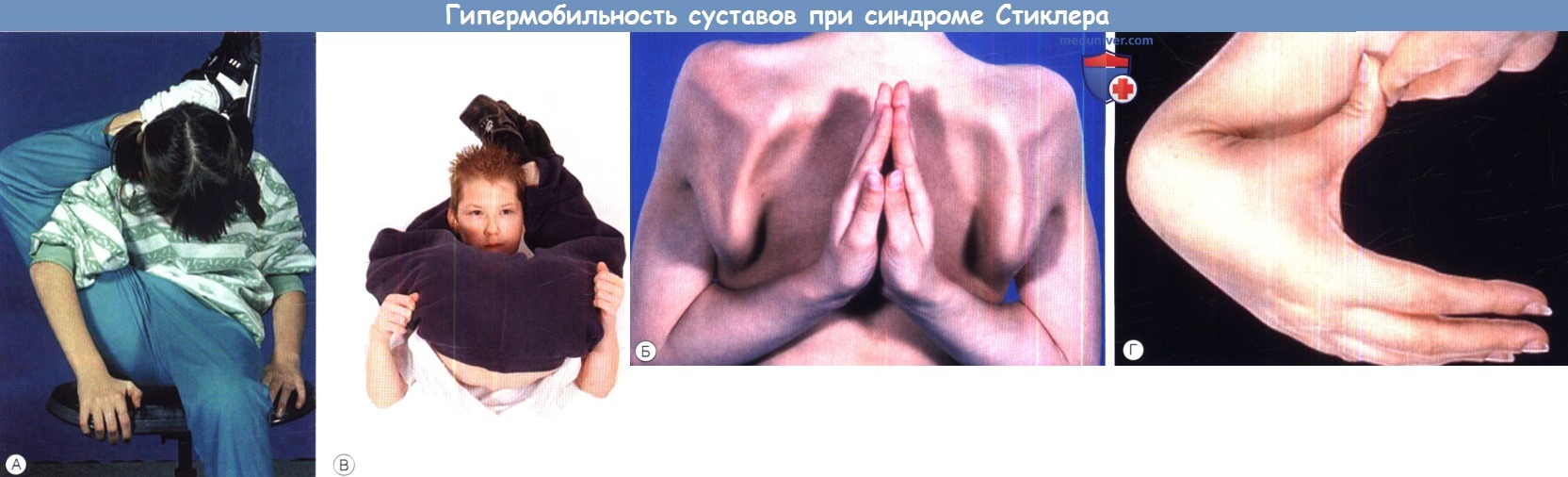 Синдром Стиклера. Гипермобильность суставов оценивается различными способами.
Синдром Стиклера. Гипермобильность суставов оценивается различными способами. 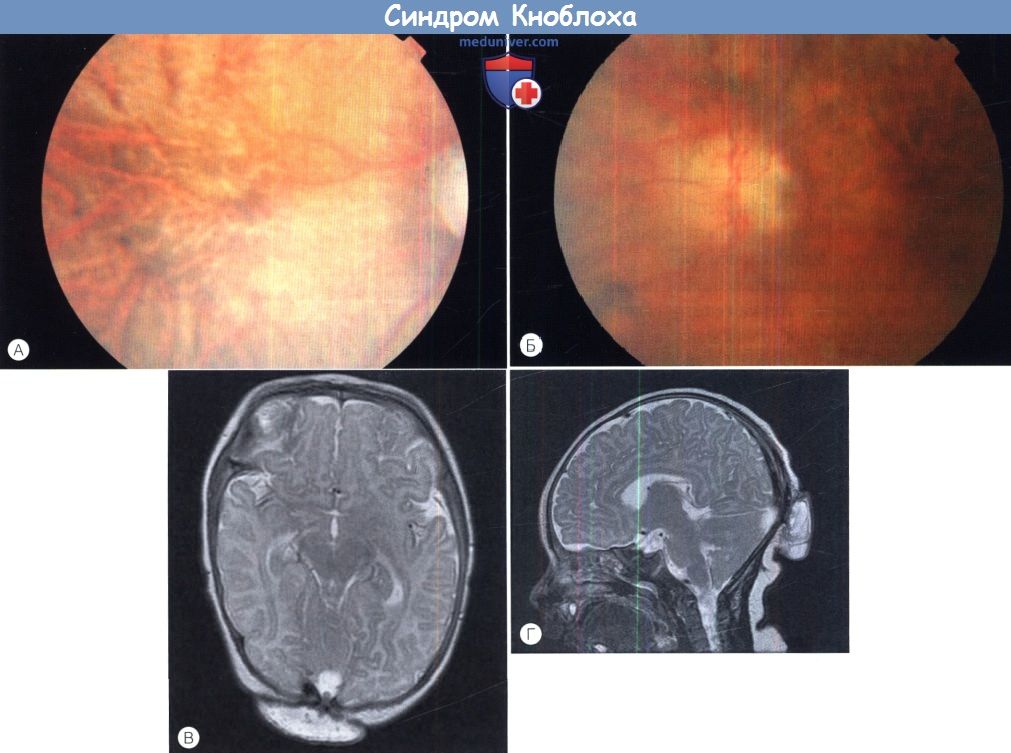 Синдром Кноблоха.
Синдром Кноблоха.
Отслойка сетчатки макулярной области левого глаза на фоне высокой близорукости в сочетании с затылочным энцефалоцеле.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021